Key Issues of Vascular Pathophysiology
Axel R. Pries
Ivo Buschmann
Helmut Habazettl
Hemodynamic Relevance of Stenoses
Hemodynamic Relations
Atherosclerotic lesions in conduit arteries may lead to a significant reduction in tissue perfusion, and consequently in the availability of oxygen. However, for ischemia to occur, the reduction in vessel diameter must be quite substantial.1,2,3
The hemodynamic consequences of vascular stenoses can be predicted using the laws of Hagen-Poiseuille (also called Poiseuille’s law) and Kirchhoff. According to Hagen and Poiseuille, the flow (Q) through a tube with a given length is proportional to the fourth power of the tube radius (r):
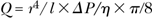 where l is the tube length, ΔP the driving pressure drop across the tube, and η the viscosity of the fluid. Equivalent is a consideration of flow resistance R = Δ P/Q:
where l is the tube length, ΔP the driving pressure drop across the tube, and η the viscosity of the fluid. Equivalent is a consideration of flow resistance R = Δ P/Q:
Thus, a decrease in radius or diameter by 50% will lead to flow reduction of nearly 94% (to 1/16) or an increase in flow resistance by 1500%. However, in contrast to vascular tone, stenoses usually only affect shorter sections of conductance vessels. For serial arrangements of vessels or vascular sections, Kirchoff’s second law indicates that the summed resistance of the serial segments (Ri) gives the total resistance of the respective flow pathway (R∑):
The pressure drop across a given section of such a flow pathway (relative to the overall pressure drop) correlates to the share of flow resistance in the respective section:
The changes in flow resistance and perfusion due to arterial stenoses are accompanied by changes in intraluminal pressure (Fig. 2-1). In healthy vascular beds, the majority of the flow resistance and thus the main pressure drop resides in the smallest arteries and arterioles (see Fig. 2-2). During development of a severe stenosis, the resistance of the affected section and the pressure drop across it (transstenotic pressure gradient) may become significant. In healthy normal vasculature, the pressure drop from large central arteries to organ arteries is very low, irrespective of the distances traveled to the particular organ. The same is true for the venous drainage with an inverse pressure gradient. As first reported by Poiseuille,4,5 the main pressure drop and flow resistance reside in the vessels of the terminal vascular beds.6,7 Here they are located predominantly in small arteries and arterioles. For the coronary circulation, the pressure drop based on in vivo measurements is shown in Figure 2-2 for the resting state and after pharmacological dilatation.8,9
On maximal loading or maximal dilatation, the flow resistance in the microcirculation decreases, substantially increasing the relative importance of flow resistance in the larger feeding and draining vessels. This interplay between increasing conduit vessel resistance with increasing severity of a stenosis and concomitantly decreasing peripheral resistance is evidenced by corresponding pressure changes along the vascular bed toward the periphery.
The presence of an arterial stenosis affects the local pressure and pressure profile along the vascular bed well before significant changes in perfusion are observed. Because of the compensatory dilatation in the peripheral small arteries and arterioles, overall resistance of the subtended vascular bed can be maintained at a near normal level for a prolonged period of time. However, this compensation is associated with a shift in the relative contribution to resistance and thus of local pressure drop from the microcirculation to the stenotic region. This leads to lower poststenotic pressure levels, even at resting conditions.
The presence of an arterial stenosis affects the local pressure and pressure profile along the vascular bed well before significant changes in perfusion are observed. Because of the compensatory dilatation in the peripheral small arteries and arterioles, overall resistance of the subtended vascular bed can be maintained at a near normal level for a prolonged period of time. However, this compensation is associated with a shift in the relative contribution to resistance and thus of local pressure drop from the microcirculation to the stenotic region. This leads to lower poststenotic pressure levels, even at resting conditions.
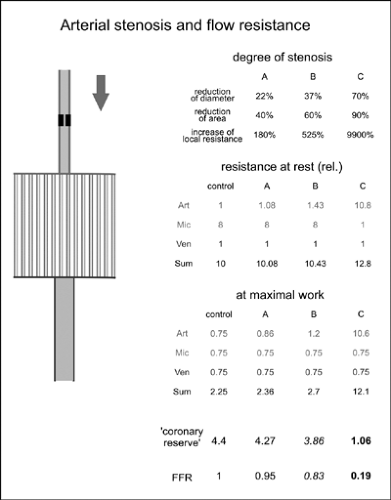 FIGURE 2-1. Distribution of resistance to blood flow for a simplified vascular bed (left) which may represent, for example, the coronary vascular bed. An artery (Art) supplies the terminal vascular bed and microcirculation (Mic), which is drained into a vein (Ven). Three different stages (A,B,C) of a single arterial stenosis, extending over 10% of the arterial length, are considered (upper right table). The tables give the relative flow resistance for the different vascular compartments for control conditions and for maximal loading. At rest, an increase in arterial resistance due to stenosis can be compensated by vasodilatation in the smaller vessels. Thus only severe stenoses lead to an increase in overall resistance. In this condition, no further dilation of the peripheral vascular bed during loading is possible, and the flow at maximal loading (or during pharmacological dilatation) is not significantly higher than in the resting state. This is reflected in the coronary reserve (flow at maximal loading/resting flow or resting resistance/resistance at maximal loading) which is close to 1, and the low values of fractional flow reserve (FFR, the ratio of the flow at maximal loading with stenosis to the flow at maximal loading without stenosis). |
Severity of Stenoses
According to the hemodynamic relations described earlier, pressure measurements exhibit a higher sensitivity than measurements of perfusion in determining stenoses, especially those of low to medium severity (Fig. 2-3). Accordingly, the pressure gradient across a stenosis measured in the catheterization laboratory with a pressure wire may be used to assess the hemodynamic severity of a stenosis. In clinical practice, this relation is utilized, for instance, in the determination of fractional flow reserve (FFR)10,11 By definition, the FFR represents the ratio of maximal myocardial flow in a patient with a suspected stenosis to the hypothetical maximal myocardial flow in the absence of a stenosis. FFR is estimated as the ratio of distal coronary pressure to aortic pressure obtained simultaneously during maximal vasodilatation. This calculation assumes that the vessel section investigated does not exhibit a significant pressure drop if no stenosis is present and that the venous outflow pressure is about zero.
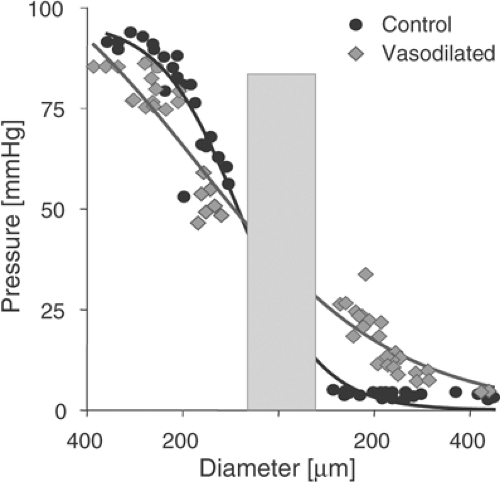 FIGURE 2-2. Intraluminal pressure drops along the vascular tree most prominently in small-resistance arteries <200 μm in diameter and in arterioles. In this example, the pressure distribution was measured in the coronary circulation during control conditions and after maximal vasodilatation with dipyridamole. Vasodilatation redistributes relative resistance away from small-resistance arteries and arterioles, resulting in pressure increase in precapillary arterioles, capillaries, and venules. The shaded area represents the segment of smallest vessels where little direct evidence is available, because of methodological difficulties in investigating this segment of the coronary microcirculation. (Adapted from Chilian WM, Layne SM, Klausner EC, et al. Redistribution of coronary microvascular resistance produced by dipyridamole. Am J Physiol. 1989;256: H383-H390. ) |
If, for example, the pressure across a stenotic segment declines by 50% of the aortic pressure (e.g., pressure distal to the stenosis would equal 50 mm Hg at an aortic pressure of 100 mm Hg), this segment would contribute 50% to the total resistance, R∑. Thus, without the stenosis, R∑ would be halved and flow would be doubled, and the corresponding FFR is resulting in FFR of 0.5 (50 mm Hg/100 mm Hg). It has been shown that for a normal myocardial function, an FFR value of 0.75 represents a valid cutoff between stenoses without (FFR >0.75) and those associated with (FFR <0.75) myocardial ischemia.
For clinical use, determination of FFR can be complemented by the measurement of the coronary flow reserve (CFR, or “flow reserve” in other organs), which is calculated by dividing the antegrade flow at maximal loading or on maximal dilatation by the antegrade flow under baseline resting conditions.
Whereas FFR is specific to the hemodynamic effects of a stenosis, CFR includes effects of flow resistance in all sections of the vascular bed, including the microvascular bed. Therefore, a successful endovascular intervention should return FFR to near 1, and the measurement of FFR allows the effect of individual measures to be judged. On the other hand, FFR is insensitive to problems downstream from the investigated section of the vasculature. Abnormal CFR may be caused, in part or in total, by increased flow resistance in the microvascular compartment. The microvascular contribution would not be addressed by the intervention and, even at normalized FFR values, symptoms may remain.
Whereas FFR is specific to the hemodynamic effects of a stenosis, CFR includes effects of flow resistance in all sections of the vascular bed, including the microvascular bed. Therefore, a successful endovascular intervention should return FFR to near 1, and the measurement of FFR allows the effect of individual measures to be judged. On the other hand, FFR is insensitive to problems downstream from the investigated section of the vasculature. Abnormal CFR may be caused, in part or in total, by increased flow resistance in the microvascular compartment. The microvascular contribution would not be addressed by the intervention and, even at normalized FFR values, symptoms may remain.
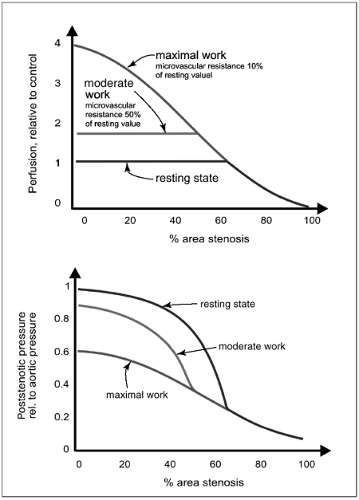 FIGURE 2-3. Perfusion and poststenotic pressure estimated for a hypothetical coronary vascular bed for resting control conditions (lower line), and for moderate (upper line) and maximal (oblique line) loading. At rest, the effect of a stenosis on perfusion (upper panel) is counteracted by a vasodilatation of small-resistance arteries and arterioles distal to the stenosis. Above a certain level of stenosis (here, reduction of vessel cross section by ∼63%), vasodilatation is maximal and resting blood flow is compromised. During loading, the decrease of peripheral resistance allows a maximal increase in perfusion to about 4 times the resting value in the absence of stenosis (coronary flow reserve). In the presence of stenosis, the dilatory capacity is (to varying degrees) already used under control conditions at the expense of the possible blood flow increase during loading. Thus, the effect of stenosis on perfusion may be masked by compensatory distal dilatation and can be uncovered during loading. In contrast, the presence of a stenosis even of lower grades is evidenced by a decline in poststenotic pressure levels (lower panel). The pressure drop across the stenosis and thus the poststenotic pressure reflects the increase in proximal resistance due to the stenosis as well as the decrease of distal resistance due to compensatory dilatation. |
Because of the fourth-power relation between diameter and flow resistance or pressure drop, measurement errors in diameter may lead to much larger errors in estimating the hemodynamic relevance of a stenosis from vascular images. Thus, such estimates bear a much higher degree of uncertainty than direct measurement of FFR and CFR. The possibility of substantial errors is especially high if vessel projections in only one direction are considered for calculations, assuming a circular cross-sectional vessel area. Better results may be expected from tomographic techniques, which allow a 3D reconstruction of the target vessel section. This spatial target vessel reconstruction may then serve as a template for flow resistance estimation using fluid dynamic simulation software.
Figure 2-4 schematically shows the dependence of functional impairment with respect to relative perfusion reserve on the severity of arterial stenoses.12 Two parameters are given, the relative perfusion reserve, that is, the flow reserve in the presence of a stenosis divided by that in the absence of the stenosis of the same vessel or in a different vascular bed used as a healthy unaffected control region,12 and the FFR.11 Significant reductions in relative perfusion reserve are observed if vessel diameter is decreased by more than about 40% or cross-sectional luminal area by more than about 60% in this model.
Microvascular Compensation
As shown in Figure 2-1, the decrease in cross-sectional luminal area caused by an arterial stenosis must be about 90% for it to result in the amount of increase in flow resistance necessary to cause reduced perfusion at rest. The main explanation for this long latency before a stenosis becomes hemodynamically significant at rest is the relatively low contribution of larger conduit arteries to overall flow resistance. Depending on the respective organ, about 5% to 15% of overall flow resistance may reside in arterial vessels with diameters ≥500 μm. Therefore, only severe narrowing of the conduit vessels will significantly increase the overall organ resistance to eventually reduce the flow.
In addition, compensatory vasodilatation in the terminal vascular bed, and specifically in the microcirculation, may mask the hemodynamic effects of a stenosis at rest. For the highest degree of stenosis in Figure 2-1, dilatation in the microvascular compartment can almost compensate even a 90% reduction in cross-sectional area in the supplying artery. Obviously, whether dilatation of small arteries and arterioles can provide this compensation depends on the resting tone in the specific vascular bed. Therefore, the impact of an arterial stenosis of a given degree will vary in different organs. Organs with a high flow reserve, such as the heart and skeletal muscle, have a high resting vascular tone. Thus, in these organs, even severe stenoses may not lead to symptoms at rest because the range for compensation is large, due to the ability of the resistance vessels to reduce the tone, that is, vasodilate. In contrast, perfusion in the kidney is near maximal at rest, indicating a low vascular tone, and the possibility for vascular compensation in the resistance compartment very limited. Typically, the flow reserve range is about 4 to 5 in the heart, up to 20 in the skeletal muscle, but only about 2 in the kidney.
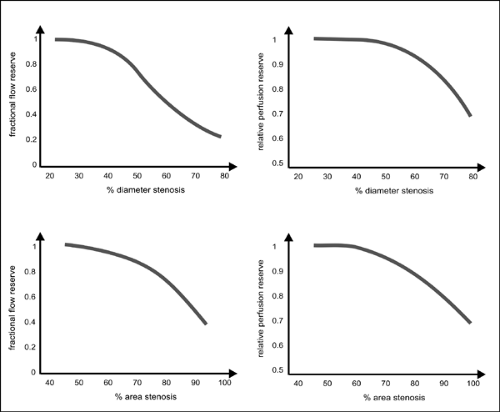 FIGURE 2-4. Fractional flow reserve, that is, the ratio of flow at maximal loading with stenosis to the flow at maximal loading without stenosis, and relative perfusion reserve, which is the ratio of blood flow at maximal loading or dilatation in regions affected by a stenosis to blood flow in unaffected control region, as a function of severity of an arterial stenosis according to results of angiographic (upper left panel; Bartunek J, Sys SU, Heyndrickx GR, et al. Quantitative coronary angiography in predicting functional significance of stenoses in an unselected patient cohort. J Am Coll Cardiol. 1995;26:328-334. ), intravascular ultrasound (lower left panel;Briguori C, Anzuini A, Airoldi F, et al. Intravascular ultrasound criteria for the assessment of the functional significance of intermediate coronary artery stenoses and comparison with fractional flow reserve. Am J Cardiol. 2001;87:136-141. ) and PET (right panels;Goldstein RA, Kirkeeide RL, Demer LL, et al. Relation between geometric dimensions of coronary artery stenoses and myocardial perfusion reserve in man. J Clin Invest. 1987;79:1473-1478 ) studies. |
During exertion, the regulatory vessels of the terminal vascular bed and microcirculation are already dilated under normal physiological conditions without arterial stenosis. Therefore, if a stenosis is present, only limited compensatory dilation is possible and the maximal perfusion during exertion (or on pharmacological dilatation) is consequently reduced. In the presence of an extremely high-grade stenosis, the compensatory peripheral dilatation in the microcirculation may be complete. A similar effect is seen with a reduction in systemic pressure. The local regulatory mechanisms, e.g., the myogenic response (see the later section entitled “Microcirculation”) lead to a compensatory decrease in tone and flow resistance aimed at maintaining resting perfusion. If a prevailing stenosis has already led to peripheral dilatation, the tolerance for an additional reduction in systemic pressure is reduced.
Collateral Circulation (Arteriogenesis)
A Natural Rescue Mechanism Providing Protection from Peripheral Hypoperfusion
Besides depending on differences in the dilatory reserve of resistance vessels in different organs, the effects of arterial stenoses also depend on the availability of alternative flow pathways to the
tissue supplied. In most organs, only peripheral regions of an area supplied by a stenosed artery may be supplied by neighboring arteries (Fig. 2-5). However, it has been shown that small arteriolar connections may develop into larger vessels, able to provide substantial blood flow. The transstenotic pressure gradient and pressure differences between arteries in the poststenotic regions and neighboring unaffected vessels has also been postulated as the main mechanism initializing growth of such collateral vessels.
tissue supplied. In most organs, only peripheral regions of an area supplied by a stenosed artery may be supplied by neighboring arteries (Fig. 2-5). However, it has been shown that small arteriolar connections may develop into larger vessels, able to provide substantial blood flow. The transstenotic pressure gradient and pressure differences between arteries in the poststenotic regions and neighboring unaffected vessels has also been postulated as the main mechanism initializing growth of such collateral vessels.
The stenosis or occlusion of an arterial vessel—as described in the previous sections—was a key issue in vascular medicine in the last century and continues to be one today. Coronary heart disease and other vascular diseases such as cerebrovascular and peripheral artery disease are likely to become even more prominent as demographic shifts and progressively unhealthy lifestyles occur in the future. From the evolutionary point of view, the massive increase in atherosclerosis, resulting from a mismatch between calorie intake and consumption, simply overeating, and aggravated by risk factors such as lack of physical exercise, smoking, or diabetes, has taken place rather recently. This might explain why evolutionary pressure has been too short to provide compensation mechanisms to cope with this disease and its devastating human costs.
One important protective endogenous mechanism against the downstream effects of arterial stenosis is the recruitment of collateral arteries and their transformation from resistance arteries into conductance arteries (arteriogenesis).13,14 However, the time course required to enlarge pre-existing collateral vessels might be too slow in many cases to allow hemodynamic compensation of rapidly evolving stenoses, resulting in perfusion deficits, ischemic endorgan damage, and—in extreme cases—peripheral tissue infarctions. Nevertheless, arteriogenesis is an important natural endogenous rescue mechanism to compensate for the reduction in arterial inflow and is briefly discussed in this section. Several experimental studies in recent years have contributed to a better understanding of the basic physiological and molecular principles of arteriogenesis.
In this context it is important to distinguish between arteriogenesis and angiogenesis. Arteriogenesis, the mechanism responsible for collateralization, represents the enlargement and remodeling of pre-existing small arteriolar collaterals between the perfusion territories of functional endarteries into conduit arteries. The major stimuli for this remodeling are mechanical forces such as shear stress and mechanical stretch. Angiogenesis describes the de novo formation of capillaries by sprouting or intussusception. The major stimuli for angiogenesis are hypoxia, such as occurs in tumor growth, or inflammation in wound healing.
The existence of collateral arteries has been controversially discussed for many years. Several investigators have described myocardial arteries as functional endarteries, while others have provided convincing data about the presence of arteriolar vessels that interconnect adjacent vascular territories in the form of networks or arcades.13,15,16
In 1956, Baroldi et al.17 demonstrated the presence from birth of mostly corkscrew-shaped collateral arteries in normal human hearts, with a luminal diameter of 20 to 350 μm and lengths ranging from 1 to 5 cm. In the hearts of patients suffering from coronary artery disease, autopsy showed that the number of coronary collaterals was increased, notably in cases with a long history of slowly evolving coronary obstructions, whereas barely any collateral vessels were found following acute myocardial infarcts. Baroldi et al. suggested that functional coronary collateral circulation results from the remodeling of vessels present in normal hearts. Indeed, in 1964, Fulton13 was able to show at a postmortem examination that the number of large coronary collaterals increased with the duration of the history of angina. When the sum of luminal diameters measured was translated into the capacity of these vessels to transport blood, it could be shown that the functional importance of a few large channels was much greater than that of a large number of small collateral channels. These observations served as an important point of departure for Schaper’s experimental studies in dogs and rabbits,14,18,19 heralding a new era of “arteriogenesis,” the positive remodeling of pre-existing collateral pathways and channels. Following convincing research data, the concept of arteriogenesis has been generally accepted.
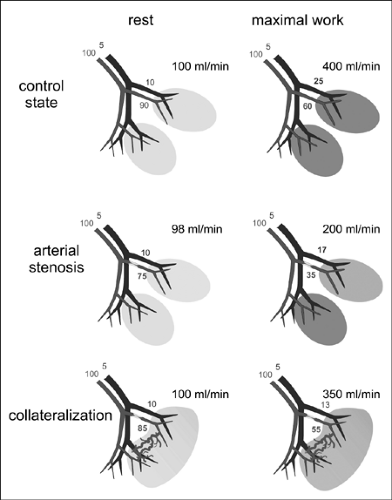 FIGURE 2-5. Schematic representation of pressure and flow distribution in a coronary vascular bed at rest and during maximal loading. Shown are typical values for normal conditions, on arterial stenosis, and after collateralization. |
Under normal conditions, collateral arteries function as a means of efficient blood flow distribution, acting as capacitors for blood displacement in nonsynchronously contracting muscles. Stenosis of a conduit artery induces a pressure fall in the poststenotic downstream vascular bed due to the pressure drop across the stenosis and compensating vasodilatation of poststenotic resistance arterioles, as indicated in Fig. 2-5. Pressure in adjacent vascular beds perfused by nonstenotic arteries remains unchanged and, thus, a pressure gradient
develops along pre-existing small collateral vessels connecting the unaffected and the poststenotic vascular territories. This pressure gradient increases flow through the collateral vessels and thus shear stress and mechanical stretch, the initial stimuli of arteriogenesis.
develops along pre-existing small collateral vessels connecting the unaffected and the poststenotic vascular territories. This pressure gradient increases flow through the collateral vessels and thus shear stress and mechanical stretch, the initial stimuli of arteriogenesis.
Taken together, the following key pathogenetic steps are important during early phases of arteriogenesis:
The pressure drop across an arterial stenosis and the recruitment of collateral pathways with a consecutive increase in shear forces across these collaterals
The up-regulation of cell-adhesion molecules on the collateral arterial endothelium (in particular intercellular adhesion molecule 1, ICAM-1)
The invasion of circulating mononuclear cells into the perivascular tissue of collateral arteries
The proliferation of smooth muscle cells (SMCs) and adventitial cells in the vessel wall, resulting in a positive outward remodeling of pre-existing collaterals (arteriogenesis)
The Role of Mechanical Forces
The mechanical forces acting on the endothelium within newly recruited collateral arteries (see previous section) trigger a cascade resulting in endothelial activation and vessel growth. As a result of increased levels of shear forces, endothelial cells (ECs) open their chloride channels, which results in a swelling and a volume increase secondary to an influx of free water.20 Cell-to-cell contacts within the coherent endothelium of the arteriolar structures are partially damaged. At this point, inflammatory cells adhere to the endothelium and transmigrate into the walls of the collateral channels.21 Currently it is believed that the endothelium is an important source for the cytokine production necessary to recruit further mononuclear cells to the site of vascular growth and proliferation by forming colony-stimulating factors, vascular endothelial growth factors, and transforming growth factors. In this context, Hoefer et al.22,23 could show that monocytes do indeed appear to play a crucial role during arteriogenesis. In their study, New Zealand White rabbits received phosphate-buffered saline (PBS), monocyte chemoattractant protein-1 (MCP-1), interleukin-8 (IL-8), neutrophil-activating protein-2 (NAP-2), or lymphotactin (Ltn) using osmotic minipumps after unilateral femoral artery ligation. Arteriogenesis was evaluated by angiography and collateral conductance measurements using fluorescent microspheres. Quantitative immunohistology was used to quantify transmigrated leukocyte subtypes after infusion of the factors. The data provided clear evidence that MCP-1 infusion attracted monocytes and granulocytes, whereas IL-8 attracted all three cell types, although monocytes were attracted to a significantly lower degree than by MCP-1. NAP-2 and Ltn selectively attracted granulocytes and lymphocytes, respectively. Importantly, among the tested cytokines, only MCP-1 stimulated arteriogenesis, as assessed by collateral conductance measurements, whereas IL-8, NAP-2, and Ltn had no significant effect on arteriogenic growth.
In a second set of experiments, in vivo treatment with monoclonal antibodies against ICAM-1 completely abolished the stimulatory effect of MCP-1 on collateral arterial growth, suggesting that the mechanism of the MCP-1-induced arteriogenesis proceeded via the attraction of monocytes to the endothelial sites, rather than by the action of the MCP-1 molecule itself. Furthermore, mice with defective selectin interactions (FT4/7-/-) did not show any significant difference in arteriogenesis, whereas ICAM-1 and Mac-1 double-knockout mice had significantly less arteriogenesis than matched controls. These results seem to indicate that ICAM-1/Mac-1-mediated monocyte adhesion to the endothelium of the preformed collateral arteries represents an essential step for arteriogenesis proceeding via selectin interaction independent mechanisms. However, further studies are needed to explore fully the molecular and cellular biology of collateral vessels’ growth.
Remodeling of Resistance Arterioles Into Conductance Vessels
The influx of circulating monocytes into the walls of the pre-existing collateral vessels leads to a cascade of molecular events resulting in a marked increase in proliferative activity and growth. Pre-existing arteriolar collaterals have a diameter of ∼50 μm. They present with one to two layers of SMCs and are morphologically indistinguishable from normal arterioles. The stages of arteriogenesis consist of (a) arteriolar thinning and (b) transformation of SMCs from the contractile into the proliferative and synthetic phenotype, followed by (c) EC and SMC proliferation, SMC migration, and formation of a neointima. Early in this process, the production of ICAM-1 and vascular cell adhesion molecule (VCAM-1) is up-regulated in ECs, and this is accompanied by accumulation of blood-derived macrophages. Moreover, an activation of gelatinase A associated with increased tissue plasminogen activator (tPA) expression and induction of gelatinase B, matrix metallopeptidase-1 (MMP-1), extracellular matrix (ECM) elastin, collagen, and proteoglycans as well as suppression of tissue inhibitors of metalloproteinases (TIMP) levels are also observed.24
In short, these multiple cellular and molecular activities during the remodeling phase of collateral artery growth are necessary to loosen up the “old” structure of the vessel including the lamina elastica and to create space for the growth of the vessels. Increased mitotic and secretory activities associated with arteriogenesis result in outward remodeling of an arteriole into an artery, which increases the original diameter of the arteriole up to 12-fold. Hence these vessels are predestined for collateral blood flow bypassing the stenosis or occlusion and providing the required oxygen and nutrition to the endangered peripheral tissue.
Therapeutic Arteriogenesis
In studies of peripheral circulation in an animal model, our group could show that certain cytokines accelerate the speed of arteriogenesis. The design of these studies was derived from Schaper’s observation identifying increased levels of monocytes on the surface of dog coronary collateral endothelium.19 In our model, ligation of the femoral artery in New Zealand White rabbits under normal conditions leads to the development of natural adaptive arteriogenesis. After 1 week of ligation, collaterals are indeed detectable, although the degree of maturation is rather low in this model. Infusing proarteriogenic factors into the collateral circulation (intra-arterially), however, leads
to a dramatic increase in collateral diameter and hence peripheral perfusion. Granulocyte macrophage colony-stimulating factor (GM-CSF) and MCP-1 in particular have been shown to be strong arteriogenesis-enhancing factors,14,25,26,27,28 in an ischemic brain model as well (Fig. 2-6). This observation has led to several experimental strategies to therapeutically enhance the collateral circulation using proarteriogenic compounds. A variety of physiological molecules have been identified that appear to promote angio- and arteriogenesis. Most act by stimulating migration and proliferation of ECs or SMCs, such as the family of fibroblast growth factors (FGF) and vascular endothelial growth factors (VEGF). Both cause vasodilatation by stimulating the release of nitric oxide (NO). The dilatory effect of these growth factors makes it, therefore, important in both animal and clinical studies to differentiate between improved perfusion caused merely by vasodilatation and true collateral growth.
to a dramatic increase in collateral diameter and hence peripheral perfusion. Granulocyte macrophage colony-stimulating factor (GM-CSF) and MCP-1 in particular have been shown to be strong arteriogenesis-enhancing factors,14,25,26,27,28 in an ischemic brain model as well (Fig. 2-6). This observation has led to several experimental strategies to therapeutically enhance the collateral circulation using proarteriogenic compounds. A variety of physiological molecules have been identified that appear to promote angio- and arteriogenesis. Most act by stimulating migration and proliferation of ECs or SMCs, such as the family of fibroblast growth factors (FGF) and vascular endothelial growth factors (VEGF). Both cause vasodilatation by stimulating the release of nitric oxide (NO). The dilatory effect of these growth factors makes it, therefore, important in both animal and clinical studies to differentiate between improved perfusion caused merely by vasodilatation and true collateral growth.
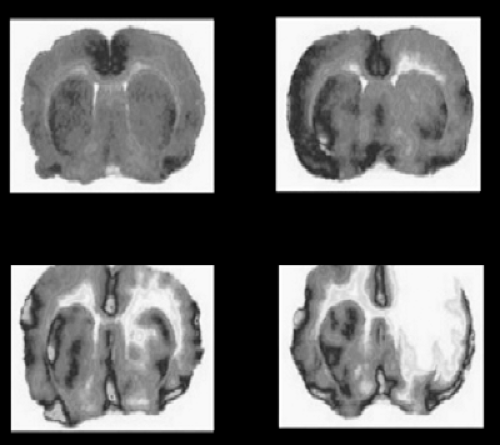 FIGURE 2-6. Protection of brain tissue by therapeutic stimulation of angiogenesis with granulocyte-macrophage colony-stimulating factor (GM-CSF). Shown is the situation under control (upper panels) and after induction of hypoxic stroke (lower panels) with (left) and without (right) application of GM-CSF. The infarct size is substantially reduced on GM-CSF treatment. |
Meanwhile several compounds have been tested experimentally and clinically to promote the growth of collateral arteries; however, the final verdict on their clinical relevance is still awaited at the time of writing.
Material Properties of Vascular Walls
In the circulation, vessels are subjected to mechanical stresses originating from blood flow and blood pressure (cf. Fig. 2-7) as well as from mechanical deformation of the vessel walls due to bending, twisting, and outside compression. The composition of the vessel wall and its mechanical properties adapt to withstand these forces. They also strongly affect the functional properties of vessels.
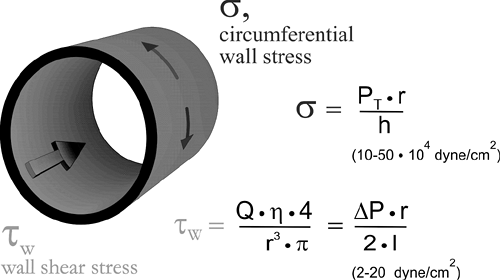 FIGURE 2-7. Mechanical forces acting on vessels (PT, transmural pressure; r, vessel radius; h, vessel wall thickness; Q, volume flow rate; η, apparent viscosity; ΔP, driving pressure difference along a vessel segment; l, length of vessel segment). Typical ranges for shear stress and circumferential wall stress (hub stress) in the microcirculation are given. |
Vessel Wall Composition
The smallest vessels, the capillaries, are composed of a layer of ECs, which in some tissues with a particularly active exchange of molecules may exhibit fenestrations or openings. The single layer of ECs is attached to a basal membrane containing mostly collagen-IV and some pericytes (see next section). Even in small arterioles and venules, the wall exhibits the typical vascular structure with three tissue layers, namely, intima, media, and adventitia. In arterioles and venules, the intima is represented by a single layer of ECs and a more or less continuous basal membrane. The media contains one to five layers of SMCs in arterioles and a sparsely developed adventitia; venules have a very thin media with fewer layers of SMCs, but a thicker adventitia containing more collagen-rich connective tissue. In larger vessels, the intimal and medial layers are thicker and more complex. In arteries, the intima exhibits a layer of connective tissue and is separated from the media by the fenestrated internal elastic membrane. Elastic membranes are also found within the media, which in addition to SMCs also contains fibroblasts and some collagen fibers. Veins again exhibit a lower amount of SMCs but more passive structural elements, mainly collagen and some elastic fibers.
In a given location within the vascular tree, veins exhibit a diameter that is approximately 1.5 to three times greater than that of the corresponding arterioles, lower wall thickness with lower relative shares of smooth muscle and elastin, but a higher collagen content. Thus, the wall-to-lumen ratio is much lower on the venular side (Fig. 2-8). For both arteries and veins the wall-to-lumen ratio decreases from small to large vessels. Typical values for normal human coronary arteries29 and carotid arteries,30,31 ranging from about 0.16 to 0.2, have been reported. As a consequence of different wall composition and different thickness, venous vessels will collapse more easily than their counterparts on the arterial side of the circulation if the transmural pressure gradient becomes negative.
Forces and Stresses
Our understanding of the mechanical properties of arterial vessels far exceeds that for veins; however, some of the general concepts may apply for both vessel classes. In situ, arteries are
under significant longitudinal tension. On detachment from the tissue and at zero pressure, they shorten by up to 40% in young experimental animals, down to ∼10% in old ones.32,33 Their unstretched length depends on transmural pressure (P). If an artery is subjected to increasing transmural pressure, a parallel increase in length and diameter is observed. However, the percentage increase in length is substantially lower than that in diameter. In experimental studies with rat arteries using a pressure increase from 50 to 150 mm Hg, percentage increases of about 80% in diameter and 30% in length have been found.32
under significant longitudinal tension. On detachment from the tissue and at zero pressure, they shorten by up to 40% in young experimental animals, down to ∼10% in old ones.32,33 Their unstretched length depends on transmural pressure (P). If an artery is subjected to increasing transmural pressure, a parallel increase in length and diameter is observed. However, the percentage increase in length is substantially lower than that in diameter. In experimental studies with rat arteries using a pressure increase from 50 to 150 mm Hg, percentage increases of about 80% in diameter and 30% in length have been found.32
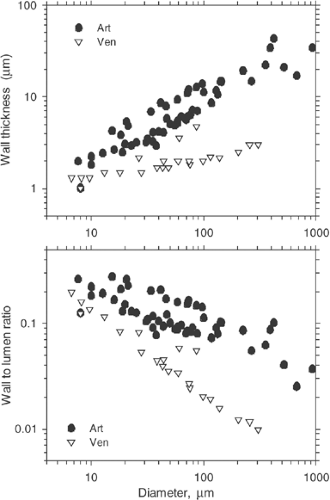 FIGURE 2-8. Relationship between wall thickness (D, top) and wall-to-lumen ratio (h/D, bottom) and vessel diameter for relaxed vessels in small laboratory animals. Wall thickness shows a relatively weak increase with vessel size, leading to a substantial decrease in the relation of wall thickness to radius. In addition, both quantities are substantially lower in venous than in arterial vessels. (Data according to Pries AR, Reglin B, Secomb TW. Structural adaptation of vascular networks: role of the pressure response. Hypertension. 2001;38:1476-1479 , andPries AR, Reglin B, Secomb TW. Remodeling of blood vessels: responses of diameter and wall thickness to hemodynamic and metabolic stimuli. Hypertension. 2005;46:726-731. ) |
The most important mechanical factor acting on the vessel wall is the circumferential tension (T, newtons per meter) calculated according to Laplace’s law for cylindrical tubes as
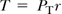 where r is the vessel radius and PT is the distending pressure. For a given structural element in the vessel wall, the tension per unit thickness (h) of the vessel wall, the circumferential wall stress or hub stress (σ; Ñ/m2 = Pa, dyne/cm2) represents the most relevant functional parameter:
where r is the vessel radius and PT is the distending pressure. For a given structural element in the vessel wall, the tension per unit thickness (h) of the vessel wall, the circumferential wall stress or hub stress (σ; Ñ/m2 = Pa, dyne/cm2) represents the most relevant functional parameter:
Mechanical Properties
Mechanical properties of the vessel wall34,35 dictate the relative increase in circumferential length (and thus diameter) or strain ε of a vessel for a given increase in circumferential wall stress. This behavior can be described by the incremental Young’s elastic modulus (E, pascals)
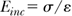 With increasing E, the vessel gets stiffer. A number of parameters are used to describe such changes in vascular mechanical properties:
With increasing E, the vessel gets stiffer. A number of parameters are used to describe such changes in vascular mechanical properties:
Compliance (C; m5/N, mL/mm Hg), i.e., the change in volume, ΔV, for a given change in pressure, ΔP:
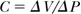
The cross-sectional compliance (Ccs; m4/N, mm2/mm Hg), where Acs is the cross-sectional area:
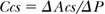
The distensibility (D; Pa-1, 1/mm Hg), where V is the vessel volume:
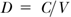
The cross-sectional distensibility coefficient (DcsC or DC; Pa-1, 1/mm Hg):
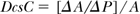
The development of techniques of measurement based on high-resolution ultrasound (≥10 MHz) combined with measurement of instantaneous blood pressure, for example, using applanation tonometry, allows the measurement of Einc, Ccs, and DcsC in a clinical setting. Typical values of Einc for human arteries range from about 200 kPa (carotid artery) to 2000 kPa (radial artery).31 Values for Ccs of about 0.07 mm2/mm Hg (carotid), and for DcsC in the range of 40 kPa-1 × 10-3 (∼300 mm Hg-1 × 10-3) (carotid) and 6 kPa-1 × 10-3 (∼45 mm Hg-1 × 10-3) (radial) have been reported.31,36 It is well established, that distensibility and compliance decrease with age and with certain chronic disease conditions, including diabetes.37
Pulse Wave
A functionally and clinically relevant circulatory parameter influenced by vascular distensibility is the pulse wave velocity (PWV, meters per second).38,39,40 PWV is related to the elastic modulus
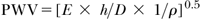
and, with some restrictions, to the inverse of compliance
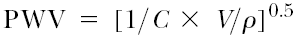 (where ρ is the specific mass of the blood), which shows that PWV increases with increasing vascular stiffness or decreasing compliance. Arterial stiffening is observed with increasing age and blood pressure, leading to a corresponding increase in PWV. For a pressure level of 100 mm Hg, PVW increases from about 6 m/sec at an age of 20 years to about 10 m/sec at 75 years. For a pressure level of 180 mm Hg, the corresponding values are 9 and 14 m/sec.
(where ρ is the specific mass of the blood), which shows that PWV increases with increasing vascular stiffness or decreasing compliance. Arterial stiffening is observed with increasing age and blood pressure, leading to a corresponding increase in PWV. For a pressure level of 100 mm Hg, PVW increases from about 6 m/sec at an age of 20 years to about 10 m/sec at 75 years. For a pressure level of 180 mm Hg, the corresponding values are 9 and 14 m/sec.
and, with some restrictions, to the inverse of compliance
Higher values of PWV lead to an earlier impact of the reflected wave on the aortic pulse pressure curve, increasing the pulse pressure amplitude (Fig. 2-9). This in turn causes increased mechanical stress on the arterial vessels and the heart. Arterial stiffness, PWV, and pulse pressure have been established as independent cardiovascular risk factors.38,39,41,42 A number of methods have been developed for waveform analysis based on noninvasive recordings on peripheral arteries (e.g., radial artery) or in peripheral vascular beds (e.g., finger) to infer aortic pressure waveforms, PVW, and additional relevant parameters of vascular mechanics.43,44 These techniques are slowly gaining a greater acceptance in clinical vascular medicine, such as for risk assessment, control of treatment, and epidemiologic evaluations.
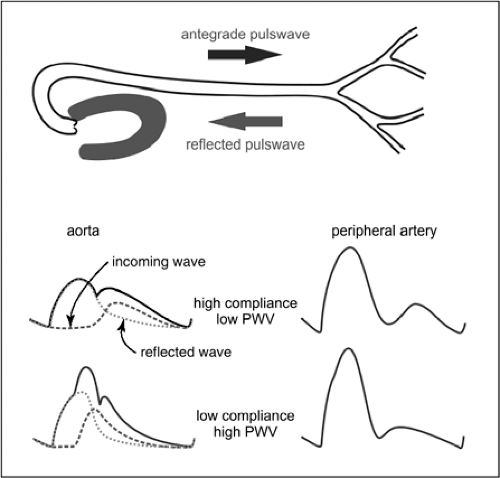 FIGURE 2-9. Pulse-wave velocity (PWV) and arterial pressure pulse. The pulse wave initiated during the ejection phase of the left heart (dotted line) travels antegrade toward the peripheral vascular beds. Here, the pulse wave is partially reflected at points of sudden impedance changes in the microcirculation (mostly vascular branch points). In persons with high vascular compliance, PWV is low, and the reflected wave (interrupted line) reaches the aorta only after closure of the aortic valve. The resulting integrative pulse pressure curve in the aorta (continuous line) consequently exhibits a secondary maximum but a relatively low maximal amplitude (pulse pressure). In contrast, with high PWV, the reflected wave hits the aorta and the left ventricle before ejection has ceased, augmenting the initial peak of the pulse curve and strongly increasing the pulse pressure amplitude. PWV also affects pulse pressure in peripheral arteries, which, because of multiple wave reflections, exhibit high pulse pressure amplitudes. |
Intravascular Interventions
The increasing number of endovascular interventions has stimulated the investigation and analysis of mechanical properties of vessels and vascular lesions.45,46,47 It has been suggested that knowledge of the mechanical properties of vessel walls and plaques may be of clinical value in the diagnosis and treatment of vascular occlusive disease. This assumption is based on the observations that composition and morphology of atherosclerotic lesions likely determine the probability of acute vascular occlusion and ischemic syndromes, as well as the responses of vascular walls and plaques, better than simple assessments of degrees of stenoses. Unstable lesions are prone to rupture followed by coagulation and thrombosis. Such lesions, in turn, exhibit a specific histological makeup demonstrating specific mechanical properties. The main histological features of unstable plaques are thought to be a large lipid pool covered by a thin fibrous cap exposed to increased stress with decreasing thickness and increasing vulnerability.
A number of techniques using both invasive (e.g., intravascular ultrasound elastography)48,49 and noninvasive (e.g., ultrasound tissue Doppler imaging of arterial wall motion)50 approaches have been used to assess the mechanical properties of defined vascular segments.
Elastography assesses local mechanical properties of tissue by applying an intraluminal pressure to which the vessel section investigated responds according to its mechanical properties. The resulting deformation of the tissue is determined using ultrasound. Using this technique, it was shown that the strain in the arterial vessel wall and in the fibrous cap of atherosclerotic lesions of various plaque types was significantly different (no plaque, fatty, fibrous, or calcified plaque).51 However, because of cumbersome measurements and calibrations, elastography has not been widely applied in clinical studies.
In the future, developments in the area of molecular medicine may lead to approaches that allow us to influence the mechanical and biological properties of vessels. Intravascular catheter interventions are an option to deliver therapeutic substances to target sections of pathologically altered vessels. Furthermore, improved knowledge of molecular mechanisms supports the development of imaging modalities, helping to locate and characterize vascular lesions.
Microcirculation
Endovascular interventions are usually directed at large to middle-sized supplying arteries, and it is not immediately clear why we should consider the microcirculation in this context. However, the reduced poststenotic pressure from upstream stenoses also affects regulatory mechanisms in the downstream microvessels. In addition, the underlying diseases that most commonly cause stenoses in the supplying arteries, such as diabetic angiopathy or atherosclerosis, are often accompanied by profound microvascular dysfunction. Also, many endovascular interventions performed to recanalize occluded or stenotic arteries to salvage ischemic tissue result in ischemia-reperfusion injury, which has long been recognized to affect downstream microvessels. Furthermore, thromboembolic complications associated with up to 30% of revascularization procedures primarily involve the downstream microvascular beds. And last,
but not least, endovascular interventions induce repetitive brief periods of ischemia and subsequent reperfusion that, if prolonged, may also affect the downstream microvessels. This section, therefore, briefly reviews the morphology and physiology of the microcirculation and its involvement in vascular disease. We focus on the microvasculature of the organs most frequently targeted by endovascular interventions, namely, heart, brain, and skeletal muscle.
but not least, endovascular interventions induce repetitive brief periods of ischemia and subsequent reperfusion that, if prolonged, may also affect the downstream microvessels. This section, therefore, briefly reviews the morphology and physiology of the microcirculation and its involvement in vascular disease. We focus on the microvasculature of the organs most frequently targeted by endovascular interventions, namely, heart, brain, and skeletal muscle.
Morphology, Angioarchitecture, and Topology
Whereas it is easy to agree that arterioles, capillaries, and venules belong to the microvascular segment of the vascular systems, their transitions and boundaries are less well defined.52 There are neither any strict morphological features nor any functional ones that allow a clear definition of the transition points from small arteries to arterioles or from venules to small veins. These transitions appear rather gradual and differ in specific vascular beds. For example, the thickness of the tunica media gradually decreases toward smaller arterial vessels until it is composed of a single layer of SMCs that terminates with the so-called precapillary sphincter in the terminal arteriole.53,54 More sophisticated approaches to assess transitions, such as the double-logarithmic plot of vessel radius versus wall thickness55 or wall stress versus vessel diameter plots,56,57,58 including large conduit vessels as well as small arterioles, each resulted in data points clustering about a single straight regression line and defying identification of a cutoff feature separating arteries from arterioles. For the purpose of this section, the microvascular segment will be considered to include all arterial vessels that contribute to blood flow control, that is, small arteries from about 200 μm in diameter to terminal arterioles. The major pressure drop has been shown to occur within this range of vessels in most tissues, including skeletal muscle, mesentery, intestinal wall, or pia mater6,7 and more recently coronary circulation.8,9,59 On the venous side, too, venules as well as small veins involved in inflammatory processes including leukocyte adhesion and migration will be included.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


