Key Issues of Vascular Pathoanatomy
Frank Kolodgie
Michael Joner
Aloke V. Finn
Elena Ladich
Robert J. Kutys
Herman K. Gold
Renu Virmani
Atherosclerotic disease persists as the most frequent cause of death in the Western world and in the 21st century and is predicted to be the primary cause of disability and death worldwide. Cardiovascular disease (CVD) is responsible for nearly half of all deaths in the developed world and for 25% in underdeveloped countries. It is predicted that by the year 2020 CVD will cause 25 million deaths annually and coronary heart disease will surpass infectious disease as the number one killer in the world.1 In this chapter we will discuss the underlying anatomy and macro- and microanatomy of the coronary arteries, their embryology, and the changes induced by atherosclerosis. The burden of diabetes and its effect on atherosclerosis as well as diabetic angiopathy will also be presented. Final discussion will include the effects of vascular interventions with both bare and drug-eluting stents on the induction and prevention of restenosis.
Development of Coronary Vessels
The initial step in the development of coronary vessels is the formation of proepicardium (PE), a transient structure that originates from the pericardial serosa in the region of the sinoatrial junction.2 The proepicardial cells are unique in that they can produce a number of different cell types during heart development. The structure of the PE varies and is dependent on the species in which it is studied. The PE is seen as a promodium. Promodia develop at the ventral midline as bilateral villous projections on the pericardial surface of the septum transversum. Each villus is composed of simple cuboidal epithelium covering a proteoglycan- and hyaluronic-acid-rich extracellular matrix (ECM) core, with occasional clusters of stellate mesenchymal cells embedded within. The villous outgrowth is the source of the epicardial layer, and the proepicardial cells are the progenitors for the coronary arteries and enter the heart around stage 17 or 18. Fate-mapping studies show that PE cells are precursors for the endothelium and smooth muscle cells of the coronary vasculature, and for the connective tissue cells that form the coronary adventitia and interstitial matrix of the myocardium.
The appearance of the epicardium and its evolving coronary vessels correlates with the transition of the heart from a primitive tube with a thin-walled epithelial-type myocardium to the thick-walled, multilayered, pumping heart found in vertebrates. The epicardial layer is closely apposed to the myocardium with a space in between that is composed of abundant extracellular matrix, rich in angiogenic factors, in which the coronary vasculogenesis is carried out. The coronary endothelial cells arise from two sources: One is the angioblasts that form elsewhere and are carried to the heart and the other is the epicardium itself. The subepicardial endothelial cells assemble into a capillarylike vessel network, and these are continuous with the sinus venosus and are considered the forerunners of the coronary venous system. The coronary vessel formation is driven by hypoxia, which is an important stimulus along with the fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) families of growth factors. Bogers et al. reported in 1989 that coronary arteries made
contact with aortic lumen not by outgrowth from the aorta as was generally accepted but rather by ingrowth of endothelial strands from the peritruncal ring of capillarylike microvessels.3 It is still poorly understood why only two stable connections are made with the aorta despite the presence of a dense collection of capillaries surrounding the aorta.
contact with aortic lumen not by outgrowth from the aorta as was generally accepted but rather by ingrowth of endothelial strands from the peritruncal ring of capillarylike microvessels.3 It is still poorly understood why only two stable connections are made with the aorta despite the presence of a dense collection of capillaries surrounding the aorta.
Basics of General Clinical Vascular Pathoanatomy
The coronary arteries, right and left, arise from the aorta close to the coronary sinus. The ostia are located in the right and left aortic sinuses and arise at the sinotubular junction at the origin of the ascending aorta. Both the coronary arteries supply the vasa vasorum of the ascending aorta. The right coronary artery arises from the right aortic sinus just above the sinotubular junction and runs in the atrioventricular groove, which separates the right atrium from the right ventricle and is surrounded by fat. The first branch of the right, the conus branch, as it arises from the right coronary sinus supplies the anterior wall of the right ventricle. The right coronary artery passes toward the inferior border of the heart and at the right lateral border gives rise to the right marginal branch that runs toward the apex of the heart. The right coronary artery curves around the right border of the heart, running in the posterior right atrioventricular groove, and at the crux of the heart gives rise to the posterior descending artery (PDA). The PDA runs toward the apex in the posterior interventricular sulcus, which separates the right from the left ventricle. At the origin of the PDA the right artery in 85% of cases gives rise to the atrioventricular (AV) nodal artery, whereas the artery to the sinoatrial node arises in 60% of cases from the right coronary artery close to its origin from the aorta. The right artery supplies the right atrium, right ventricle, and posterior portion of the interventricular septum and an adjoining portion of the left ventricular free wall.
The left main coronary artery arises from the left coronary sinus, just below the sinotubular junction, passes behind the right ventricular outflow tract reaching between the left auricle and the pulmonary trunk, and divides into two main branches, the left anterior descending (LAD) artery and the left circumflex (LCx) arteries. Rarely the left main artery is absent and there are two separate ostia for the LAD and LCx. The larger branch is the LAD, which courses along the epicardial surface of the anterior interventricular sulcus to the apex of the heart and is surrounded by fat for most of its course. The LAD branches are the septal and the diagonal branches. The septal branches arise from the LAD at 90-degree angles and pass into the interventricular septum, supplying the anterior interventricular septum of the ventricle. The diagonal branches of the LAD pass over the anterolateral wall of the left ventricle. There is a wide variation in the number and size of the diagonal branches, as most patients have one to three supplying the anterolateral wall of the left ventricle. The LAD supplies the anterior two thirds of the interventricular septum and the anterior wall of the left ventricle. The left circumflex branch follows the left atrioventricular sulcus around the left border of the heart and gives rise to left obtuse marginal branches, which supply the lateral wall of the heart. The LCx also supplies the left atrium and the left surface of the heart. The artery that gives rise to the PDA is said to be the dominant artery with the right artery being the dominant artery in 85% of cases (Fig. 1-1).
In 10% of cases the left circumflex gives rise to the PDA and therefore is the dominant artery; the right is usually a very small artery and terminates at the crux of the heart. In another 5% of cases it is a codominant blood supply with the right giving rise to PDA and the left providing all the lateral branches.
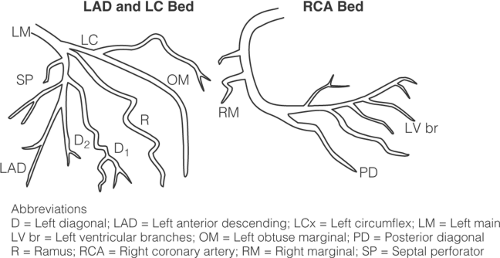 FIGURE 1-1. Coronary artery anatomy. Diagram of the right and left epicardial coronary arteries as they arise from the aorta (right dominance). The four major arteries that must be described in detail are the right (RCA), left main (LM), left anterior descending (LAD), and the left circumflex (LC) coronary arteries. Not uncommonly, severe coronary artery disease (>75% cross-sectional area luminal narrowing) affects the smaller branches (R, Ramus intermediate; LD, left diagonal; OM, left obtuse marginal; PD, posterior descending; RM, right marginal). The proximal LAD extends from the origin of LAD to the D1, the mid LAD extends from the diagonal 1 to 2.5 cm distally, and the remainder of the LAD is distal. The left circumflex is divided into proximal and region proximal to LOM, and the continuation of the LC is the distal segment. The right proximal is the first 2 cm of the artery, the mid extends up to the RM, and the distal extends up to the PDA. Occasionally, the right coronary can extend beyond the PD (so-called hyperdominant right) and provide left ventricular branches (LV br). (Reproduced with permission from Virmani R, Burke A, Farb A, et al., eds. Cardiovascular pathology. 2nd ed. Major Problems in Pathology. Philadelphia: WB Saunders, 2001:4 ). |
The left main coronary artery is usually 10 to 15 mm in length and varies in diameter from 3 to 6 mm; however, it may be as long as 2.5 cm, although this is seen in <10% of cases. The left anterior coronary artery extends from its origin to the apex of the left ventricle. At the apex the LAD wraps around the apex and supplies the posterior wall of the left ventricle. The left circumflex travels in the atrioventricular groove underneath the left atrial appendage and varies in length depending on the number of left obtuse marginal branches (LOM). Usually after the take-off of the LOM branches, the left circumflex artery tends to suddenly taper and becomes small. The right artery passes along the right atrioventricular groove and gives rise to the PDA near the crux of the heart (the meeting point of the atrioventricular groove and the interventricular sulcus). The first branch of the right artery is usually the conus branch, which arises near the take-off of the right artery in 50% of cases and in the rest arises as a separate ostium in the right coronary sinus. The second branch is usually the sinoatrial node artery, which arises from the right artery in 60% of cases; in 40% of cases it arises from the left circumflex. The posterior papillary muscle is supplied only by the right coronary artery, whereas the anterolateral papillary muscle gets a dual supply from the LAD and LCx.
The coronary arteries vary in size depending on the weight and height of the patient. Anatomic as well as IVUS studies have shown that coronary arteries following their origin from the ostia normally taper until they become intramural.4,5,6,7
Arterial tapering was determined by measuring the internal elastic lamina (IEL) area in normal arteries with minimal intimal thickening (total intimal cross-sectional area <1 mm2). The normal tapering between serial sections (approximately 10 mm between sections) was 1.20 ± 2.40 mm2 per cm for LAD and 1.20 ± 2.15 mm2 per cm for circumflex artery.8 Abnormal tapering between adjacent sections is defined as tapering > ± 2 SD of the expected tapering. Based on the foregoing study, abnormal tapering or negative vessel remodeling is present when a 25% or greater decrease in IEL is present between sections, i.e., every 1 cm. Glagov et al. first introduced the concept of arterial remodeling in the presence of atherosclerosis, in a landmark paper published in 1987; this topic will be addressed with atherosclerosis.9
The normal coronary artery is a muscular vessel with a well-defined internal and an external elastic lamina (EEL). In between these structures is the medial wall, composed of organized layers of smooth muscle cells with few interspersed proteoglycans and collagen fibers (Fig. 1-2). The intima exhibits adaptive thickening especially at branch points, consisting of smooth muscle cells, proteoglycan-rich matrix, and type III collagen fibers. Endothelial cells with well-formed junctions line the lumen surface. The outer adventitia juxtaposed to the EEL is composed of type I collagen, and its thickness is almost equivalent to media. Away from the EEL, however, the collagen fibers are loosely arranged, with interspersed mature fat. Further, present in the adventitia are fibroblasts and vasa vasorum that may or may not have surrounding smooth muscle cells (Fig. 1-2). Close to the ostia of the coronary arteries, the medial wall shows presence of multilayered elastic fibers, but these soon disappear within 1 cm of the origin of the artery. The medial wall thickness varies from 150 to 350 μm and varies with the size of the coronary artery, which also varies with the heart weight. The larger the heart, the larger the coronary artery, and as will be discussed, there is positive remodeling of the artery as atherosclerosis advances.
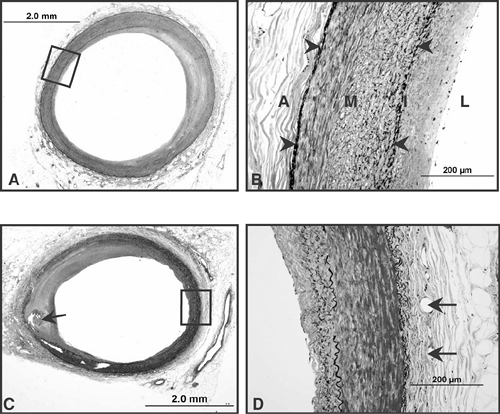 FIGURE 1-2. Coronary artery sections from patients dying a non-cardiac death in the absence of any coronary artery disease. (A) and (B) are low and high power views of the proximal LAD with mild adaptive intimal thickening. Note the high power view in (B) of the boxed area in (A) showing well demarcated (arrowheads) external (EEL) and an internal elastic lamina (IEL) however, in the media (M) towards the EEL the smooth muscle cells (SMC) are circularly arranged while those towards the IEL are tranversely arranged and interspersed in between are elastic fibers. The adventitia (A) is thin while the intima (I) consists of SMCs and proteoglycan matrix. (C) and (D) are sections from the right coronary artery (RCA) note an eccentric focally calcified neointima (arrow) with underlying markedly thinned and destroyed media. The opposite side of the artery shows minimal intimal thickening, which is seen at high power in (D). The media is demarcated by the EEL and IEL (black) on either side of the SMC rich medial wall. The intima is mildly thickened and consists of few SMCs interspersed in a proteoglycan matrix. The adventitial wall shows the presence of vasa vasorum (arrows) within the collagen rich adventitia. |
Progression of Human Coronary Atherosclerosis
The cellular and acellular components of atherosclerosis and those leading to plaque rupture reside in the vessel wall and circulating blood (Fig. 1-3). Those integral to the vessel wall include endothelial and smooth muscle cells, together with the extracellular matrix represented by proteoglycans, collagen, and elastic fibers. Proatherogenic circulating factors consist of plasma lipoproteins, fibrinogen, clotting factors, and cells: platelets, red blood cells, monocytes, lymphocytes, mast cells, and neutrophils.10 The first intimal change observed in human coronary arteries is known as adaptive intimal thickening, characterized by smooth muscle cells and surrounding proteoglycan matrix arranged in a relatively thin layer above the media. These lesions, mostly occurring at branch points, are observed in ∼30% of infants at birth, and their distribution and development correlate with the presence of atherosclerotic plaques later in life.11 Cell replication is generally low at these sites, suggesting that plaques that develop during adulthood may be clonal in origin. Tabas et al. have proposed that the extracellular matrix in early lesions contains enzymes capable of retaining lipids, a step toward early necrotic core formation.12 There are few published studies, however, on the early evolution and development of intimal mass lesions in
humans, although a clearer understanding of this concept is critical to elucidating the natural progression of the disease process.
humans, although a clearer understanding of this concept is critical to elucidating the natural progression of the disease process.
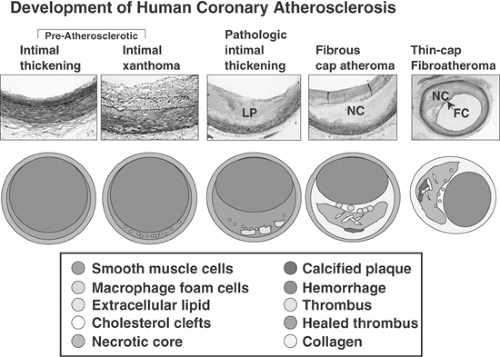 FIGURE 1-3. Development of human coronary atherosclerosis. Coronary lesions are uniformly present in all populations, although intimal xanthomas (so-called fatty streaks) are more prevalent with exposure to a Western diet. Preatherosclerotic lesions (adaptive intimal thickening and intimal xanthoma) occur soon after birth, although the latter are known to regress with age. Adaptive intimal thickening consists mainly of smooth muscle cells (SMCs) in a proteoglycan matrix, whereas intimal xanthomas contain macrophage-derived foam cells, T lymphocytes, and varying amounts of SMCs. Pathologic intimal thickening versus atheroma: Pathological intimal thickening (PIT) is a poorly defined entity, referred to in the literature as “intermediate” lesion. True necrosis is not apparent, as there is no evidence of cellular debris; lipid pools (LP) are seen deep in the lesion. The tissue over the lipid pools is rich in SMCs and proteoglycans; scattered macrophages and lymphocytes may also be present. The more definitive lesions, of fibrous cap atheroma, classically shows a true necrotic core (NC) containing cholesterol esters, free cholesterol, phospholipids, and triglycerides. The fibrous cap consists of SMCs in a proteoglycan-collagen matrix, with a variable number of macrophages and lymphocytes. The thin-cap fibroatheroma (vulnerable plaque): Thin-cap fibroatheromas are lesions with large necrotic cores containing numerous cholesterol clefts. The overlying fibrous cap (FC) is thin (<65 μm) and heavily infiltrated by macrophages; SMCs are rare and microvessels are generally present in the adventitia. (Reproduced with permission from Virmani R, et al. Arterioscl Thromb Vasc Biol. 2000;20:1262-1275. ) (See Color Fig. 1-3.) |
The first coronary lesions characterized by inflammatory cells are defined as intimal xanthomas or “fatty streaks” in the AHA classification.13 In humans, these lesions primarily consist of fat-laden macrophages intermixed with smooth muscle cells and proteoglycan matrix. Intimal xanthomas are commonly found in the thoracic aorta of young individuals, whereas advanced morphologies beyond fatty streaks in the adult thoracic aorta are usually absent.14 Other typical sites of intimal xanthomas occurring at sites where advanced lesions are usually located are the abdominal aorta and the proximal portion of the left anterior descending coronary artery. Therefore, it is our understanding that fatty streaks may not be part of the atherosclerotic process, because the vast majority regress with age.15
Coronary lesion progression from the intimal mass lesion is thought to involve a transitional plaque referred to as pathologic intimal thickening (PIT) or type III lesion in the AHA classification. These plaques are defined as acellular areas containing remnants of smooth muscle cells with extracellular lipid, lipid pools, and extensive proteoglycan matrix.15 Few viable smooth muscle cells are found among numerous PAS-positive empty shells, representing the basement membranes of earlier “viable” smooth muscle cells.16 Attenuated smooth muscle cells contain plasma membrane remnants, and apoptotic bodies are visible by electron microscopy. The lipid pools may also contain free cholesterol appearing as cholesterol clefts in paraffin sections.17 Specific stains reveal speckled granular calcification, which is underappreciated by hematoxylin and eosin staining in the area of the lipid pools. It is generally believed that smooth muscle apoptosis along with the accumulation of lipids may be responsible for the lipid pools seen in these lesions. The proteoglycan matrix within lipid pools found in PIT is rich in sulfated glycosaminoglycans, which can effectively bind apolipoprotein B. Additional proteoglycans that accumulate during this phase include dermatan sulfate, decorin and biglycan.18 Other negatively charged proteoglycans such as chondroitin sulfate function in the retention of apo B-100.19
The cellular composition above the lipid pool often contains scattered intact lipid-laden macrophages in addition to T lymphocytes.20 In contrast, B lymphocytes are restricted mostly to the adventitia; only a few are found within the developing plaque.
Similarly, mast cells may also be present, but these cells are far fewer than other cell types.21 No true necrosis is present at this stage, although these lesions do contain free cholesterol, fatty acid, sphingomyelin, lysolecithin, and triglycerides.22
The next stage of lesion development is the fibroatheroma, characterized by distinct layers of superficial fibrous tissue surrounding an area of necrotic core. The fibrous cap consists of smooth muscle cells and proteoglycan-collagen matrix, with varying degrees of inflammatory cells, mostly macrophages and lymphocytes. The fibrous cap thickness primarily distinguishes the fibroatheroma from the thin fibrous cap atheroma (classic “vulnerable” plaque).15 Moreover, we have recently subtyped fibroatheromas into “early” and “late” depending on the characteristics of the necrotic core (Fig. 1-4).23 The “early fibroatheroma” is a lesion with a lipid-rich matrix containing
proteoglycans, versican, hyaluronan, and type III collagen interspersed with intact foamy macrophages. Early necrotic cores are recognized by special proteoglycan and macrophage stains. In contrast, necrotic cores with late necrosis show numerous cholesterol clefts, cellular debris, and an absence of extracellular matrix (especially versican and hyaluronan).23 Within the center and perimeter of necrosis, ghosts of macrophages identified by anti-CD68 staining with ill-defined cell membranes are found, and picrosirius red staining within the necrotic core is negative for collagen. The thin-cap fibroatheroma (TCFA) is identified based on the finding of a necrotic core that has a fibrous cap <65 μm thick, is rich in type I collagen, and is heavily infiltrated by macrophages; lymphocytes and smooth muscle cells are rare (Fig. 1-5).24,25 It is important to classify this lesion as a distinct entity from plaque rupture, because its early diagnosis as a precursor lesion to rupture and its identification and treatment in life may help reduce the incidence of sudden coronary death and the morbidity associated with coronary heart disease.
proteoglycans, versican, hyaluronan, and type III collagen interspersed with intact foamy macrophages. Early necrotic cores are recognized by special proteoglycan and macrophage stains. In contrast, necrotic cores with late necrosis show numerous cholesterol clefts, cellular debris, and an absence of extracellular matrix (especially versican and hyaluronan).23 Within the center and perimeter of necrosis, ghosts of macrophages identified by anti-CD68 staining with ill-defined cell membranes are found, and picrosirius red staining within the necrotic core is negative for collagen. The thin-cap fibroatheroma (TCFA) is identified based on the finding of a necrotic core that has a fibrous cap <65 μm thick, is rich in type I collagen, and is heavily infiltrated by macrophages; lymphocytes and smooth muscle cells are rare (Fig. 1-5).24,25 It is important to classify this lesion as a distinct entity from plaque rupture, because its early diagnosis as a precursor lesion to rupture and its identification and treatment in life may help reduce the incidence of sudden coronary death and the morbidity associated with coronary heart disease.
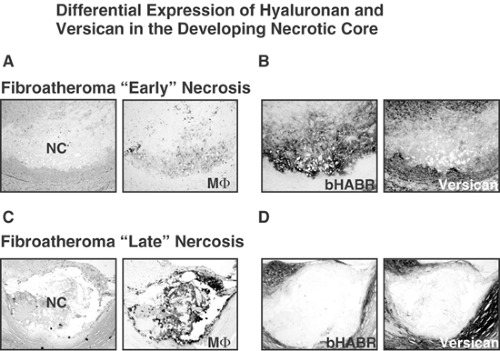 FIGURE 1-4. Fibroatheroma with early (A,B) and late (C,D) necrosis. A: Note that the early core (NC) shows an area of acellularity containing proteoglycans (Movat Pentachrome, blue-green) and small cholesterol clefts interspersed with CD68+ macrophages (MΦ). B: This same area shows intense staining for hyaluronan with moderate expression of versican. C: The late necrotic core consists of a well-defined area of cellular and acellular debris surrounded by CD68+ macrophages. D: The late core shows a virtual absence of hyaluronan and versican. (Matrix staining courtesy of Dr. Tom Wight, Hope Heart Institute, WA.) |
Lesions with Thrombi
The most frequent cause of thrombosis is plaque rupture (Fig. 1-6), occurring in 60% of cases of sudden coronary death with thrombi. Acute plaque rupture consists of a luminal platelet-fibrin thrombus continuous with an underlying necrotic core.15 The connection between the thrombus and the lipid core is through a disrupted thin fibrous cap infiltrated by macrophages. The fibrous cap is composed of type I collagen and has few or no smooth muscle cells. The second most frequent cause of coronary thrombosis is plaque erosion, which occurs in 35% of cases of sudden death with a thrombus (Fig. 1-7).15,26 Plaque erosion is defined as an acute thrombus in direct contact with the intimal plaque without rupture of a lipid core as demonstrated by serial sections. Typically, the endothelium is absent at erosion site. The exposed intima consists predominantly of smooth muscle cells and proteoglycans, and surprisingly, the erosion site contains few inflammatory cells. Plaque erosion is the most frequent cause of thrombosis in women <50 years of age, whereas in men <50 years plaque rupture is more frequent (Table 1-1). The least frequent cause of thrombosis is calcified nodule, which is present in 2% to 5% of cases.15. It is characterized by having an underlying calcified plate that is broken into multiple pieces, and calcified nodules are located on the lumen and are in direct contact with the flowing blood, which leads to platelet-rich luminal thrombus (Fig 1-6).
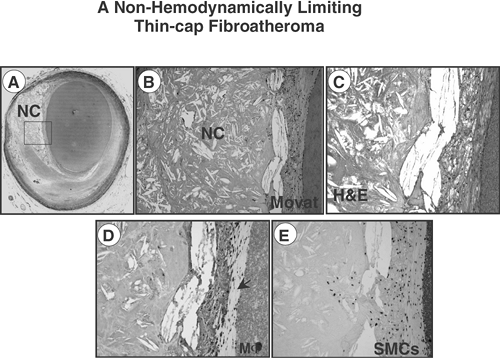 FIGURE 1-5. A non-hemodynamically limiting thin-cap fibroatheroma. A: A thin-cap fibroatheroma having a necrotic core (NC) and an overlying thin fibrous cap (65 μm). B: High-power view of the boxed area in (A). Note an advanced necrotic core with large number of cholesterol clefts with a loss of matrix containing numerous cholesterol clefts and cellular debris. The fibrous cap is heavily infiltrated by macrophages, better seen in (C) (H&E). D,E: Macrophage infiltration (CD68 positive) and rare staining of smooth muscle cells (α-actin positive) in the fibrous cap. (Reproduced with permission from Kolodgie FD, et al. Heart. 2004;90:1385-1391. ) |
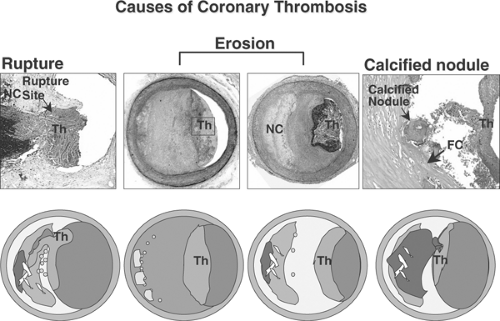 FIGURE 1-6. Atherosclerotic lesion with luminal thrombi. Ruptured plaques are thin fibrous cap atheromas with luminal thrombi (Th). These lesions usually have an extensive necrotic core (NC) containing large numbers of cholesterol crystals and a thin fibrous cap (<65 μm) infiltrated by foamy macrophages and a paucity of T lymphocytes. The fibrous cap is thinnest at the site of rupture and consists of a few collagen bundles and rare smooth muscle cells. The luminal thrombus is in direct communication with the lipid-rich core. Erosions occur over lesions rich in smooth muscle cells and proteoglycans. Luminal thrombi overlie areas lacking surface endothelium. The deep intima of the eroded plaque often shows extracellular lipid pools, but necrotic cores are not uncommon; when present, the necrotic core does not communicate with the luminal thrombus. Inflammatory infiltrate is usually absent, but if present, is sparse and consists of macrophages and lymphocytes. Calcified nodules are plaques with luminal thrombi showing calcific nodules protruding into the lumen through a disrupted thin fibrous cap (FC). There is absence of an endothelium at the site of the thrombus, and inflammatory cells (macrophages, T lymphocytes) are absent. (Reproduced with permission from Virmani R, et al. Arterioscl Thromb Vasc Biol. 2000;20:1262-1275. ) (See Color Fig. 1-6.) |
Intraplaque Hemorrhage
Intraplaque hemorrhage is a frequent event and is most often observed in patients dying from acute plaque rupture. In the early to mid-20th century, several leading pathologists forwarded the hypothesis that intraplaque hemorrhage is a major contributor to the progression of coronary atherosclerosis; however, the precise nature of this relationship was not well understood.27,28,29 Recent studies from our laboratory suggest that plaque hemorrhages are more frequent in the coronary vasculature in patients dying from rupture as compared to plaque erosion or stable lesions with >75% cross-sectional area luminal narrowing.10 In an effort to further understand the influence of intraplaque
hemorrhage on lesion progression, we examined various types of human coronary plaques for hemorrhagic events.23
hemorrhage on lesion progression, we examined various types of human coronary plaques for hemorrhagic events.23
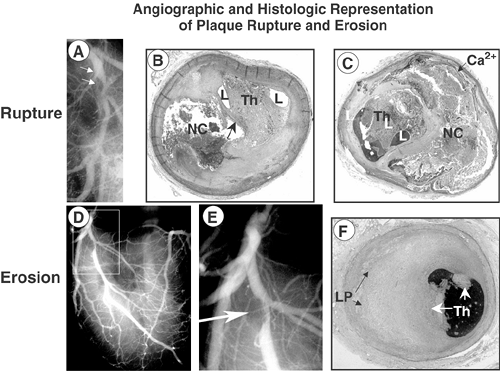 FIGURE 1-7. Angiographic and histologic representation of plaque rupture and erosion. A 43-year-old white man with no known history of risk factors was found unresponsive in the bathroom where he was last seen alive 20 minutes earlier. A: Postmortem angiogram shows the LAD at the origin of the left diagonal with a near total occlusion. Sections taken from these sites show a plaque rupture [arrow, (B)] with an underlying necrotic core (NC). The occluded artery shows an organizing thrombus with small lumens (L). C: The fibrous cap is intact with a large underlying necrotic core with peripheral calcification (Ca2+) and the lumen shows organizing thrombus (Th) with small lumens (L). At autopsy there was a healing transmural myocardial infarction present in the distribution of the LAD. Postmortem angiogram (D,E) and corresponding photomicrograph (F) from a 38-year-old man who was last seen alive 8 hours antemortem, died from sudden coronary death. A focal stenosis is present in the left anterior coronary artery (boxed area), which is highlighted in (A), and an arrow points to the area of narrowing at the take-off of the left diagonal. F: Acute nonocclusive luminal thrombus (Th) is present on the surface of an erosive plaque rich in proteoglycans (green) and the underlying plaque shows pathologic intimal thickening with lipid pools (LP). (Reproduced in part from Figure 4, Farb A, et al. Circulation. 1995;92:1701-1709. ) (See Color Fig. 1-7.) |
In a relatively large series of human coronary plaques from sudden coronary death victims, there was a greater frequency of previous hemorrhages in coronary atherosclerotic lesions prone to rupture (as detected by glycophorin A staining) relative to lesions with early necrotic cores or plaques with pathologic intimal thickening (Fig. 1-8).23
Significantly, the degree of reactive glycophorin A staining and the level of iron deposits in the plaque corresponded to the size of the necrotic core, and changes in these variables paralleled an increase in macrophage density, suggesting that hemorrhage itself serves as an inflammatory stimulus (Table 1-2).23 It is generally accepted that apoptotic macrophages are a likely source of free cholesterol in plaques; however, it is entirely feasible that free cholesterol within the necrotic core could be derived from other sources, including erythrocyte membranes. By contributing to the deposition of free cholesterol, macrophage infiltration, and enlargement of the necrotic core, the accumulation of erythrocyte membranes within an atherosclerotic plaque may represent a potent atherogenic stimulus. These factors may increase the risk of plaque destabilization.
Erythrocyte Membrane-Derived Free Cholesterol and Plaque Progression
As proof of concept, we developed an animal model of simulated intraplaque hemorrhage to assess the role of erythrocytes in lesion progression.23 The direct injection of packed erythrocytes (25 μL to 50 μL) into quiescent aortic atherosclerotic plaques produced excessive macrophage infiltration along with free cholesterol crystals, and iron colocalized to areas of red blood cells. In contrast, control (noninjected) lesions showed the characteristics of a regressed lesion with far fewer lesional macrophages and free cholesterol. Neutral lipids identified by Oil Red O were also significantly greater in plaques with injected erythrocytes when compared with controls.
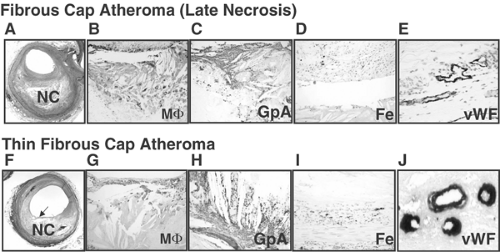 FIGURE 1-8. Late core (A-E) and thin fibrous cap atheroma (F-J) showing intraplaque hemorrhage. A: Low-power view of a fibrous cap atheroma with a late necrotic core (NC) (Movat Pentachrome × 20). B: Intense staining of CD68-positive macrophages is seen within the necrotic core. C: Extensive glycophorin A (GpA) positive erythrocyte membranes colocalized with numerous cholesterol clefts within the necrotic core (× 200). D: Iron deposits (blue) are seen within macrophage foam cells (× 200). E: Microvessels bordering the necrotic core show perivascular von Willebrand factor (vWF) deposition (× 400). F: Low-power view of a fibroatheroma with a thin fibrous cap (arrow) overlying a relatively large necrotic core (Movat Pentachrome, × 20). G: The fibrous cap is devoid of smooth muscle cells (not shown) and is heavily infiltrated by CD68-positive macrophages (MΦ, × 200). H: Intense glycophorin (GpA) staining of erythrocyte membranes within the necrotic core colocalized with cholesterol clefts (× 100). I: Adjacent coronary segment with accumulated iron (blue pigment) in a macrophage-rich region deep within the plaque (× 200). J: Perivascular diffuse deposits of von Willebrand factor in microvessels; indicates leaky vessels bordering the necrotic core (× 400). (Reproduced with permission from Kolodgie FD, et al., N Engl J Med. 2003;349:2316-2325. ) (See Color Fig. 1-8.) |
TABLE 1-1. Distribution of Culprit Plaques by Sex and Age in 241 Cases of Sudden Coronary Death | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Thus the animal studies offer further evidence that episodic hemorrhages in plaques contribute to accumulated free cholesterol and macrophage infiltration.23 The contribution of erythrocyte membrane cholesterol to necrotic core volume is predicted to be substantial because repeat intraplaque hemorrhages are thought to occur over years. Consistent with this notion, even small bleed volumes of only 0.137 μL per day whole blood (0.068 μL packed red blood cells) over a 2-year period would be sufficient to make a significant contribution to necrotic core size. Since the liquid volume of cholesterol in a single red blood cell is equivalent to 0.378 μm3, approximately 5.2 × 108 RBCs or a single bleed of 100 μL whole blood would contribute at least 0.2 mm3 or 10% of membrane-derived free cholesterol to necrotic core volume, assuming 10% exchange efficiency.30 As in internal bleeds, the bulk of the erythrocyte would be metabolized over several days, and because cholesterol cannot be metabolized internally, it would be available for absorption into the necrotic core in addition to its uptake by macrophages, which in turn presumably gives up cholesterol to the core by apoptotic cell death. In emerging serial MRI data of carotid plaques over 18 months, evidence of repeat hemorrhages is suggested to contribute significantly to necrotic core volume and lesion bulk.31 Therefore, RBC-derived cholesterol may represent a critical transition factor promoting the conversion of a stable plaque to an unstable phenotype.
TABLE 1-2. Morphometric Analysis of Plaque and Hemorrhagic Events in Human Coronary Arteries from Sudden Death Victims | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Coronary Plaque Morphology in Patients with Stable and Unstable Angina
Coronary morphology in patients with stable and unstable angina is difficult to assess because most patients with angina do not die from their disease. Patients with unstable angina who come to autopsy most likely have had an acute myocardial infarction (MI) evolving from an angina syndrome. One possible source of data regarding plaque morphology in patients with angina is from autopsy studies in patients who die during or soon after coronary bypass surgery. In the mid-1970s, Guthrie et al. published the earliest work in 35 patients with stable angina who died after coronary bypass.32 In this study, the extent of severe atherosclerosis in >95% of patients involved two or more vessels, including left main disease in approximately 30%; thrombosis was an infrequent finding. In another study, Han Gartner et al. described substantial plaque variability in the coronary lesions of patients with stable angina who died suddenly within 6 hours of onset of symptoms.33 They reported that 76% of coronary segments with >75% cross-sectional luminal narrowing showed concentric lesions, 47% were fibrous plaques, and 28% were lipid rich. The remaining 24% of severely narrowed lesions were eccentric and divided equally among fibrous and lipid-rich lesions. Recanalized segments were present in 79% of the hearts. There is no mention of the number of lesions with thrombi.
By coronary angiography, Levin et al. showed smooth borders that impart an “hourglass” configuration correlated histologically with fibrous or fatty uncomplicated plaques.34 Angiographic irregular borders or intraluminal lucent areas were found primarily in complicated plaques with rupture. In subsequent angiographic studies, Ambrose et al. reported a higher frequency of type II eccentric lesions (asymmetric with a narrow neck, irregular borders, or both) in patients with unstable angina compared with those with stable angina.35
The lesion progression in stable angina is greatest at angiographic sites with complex versus smooth stenosis and occurs in 16% of complex lesions but not in smooth lesions at an interval of 9 months (range 3 to 24 months). The annual increase in plaque growth has been shown to be 11.4 ± 28% and 1.5 ± 14% for complex and smooth lesions, respectively (p <0.01).36 We have shown that plaques with no prior ruptures versus those with ruptures have an increased luminal narrowing, and only 11% of culprit plaques show no prior rupture, thus suggesting that repeated ruptures are the mechanism by which plaques increase in size with eventual severe narrowing.37 haracterization of culprit lesions by angioscopy in various coronary syndromes reveals the different mechanisms of ischemia. The predominant lesion in acute myocardial infarction is an ulcerated, yellow plaque with thrombus. In unstable angina, different substrates can be seen, from the lipid-rich lesion with thrombus to the fibrous smooth plaque, reflecting a varied physiopathology.38
We have reported our findings in 450 cases of sudden coronary death in blacks (n = 130) and whites (n = 320), mean age 51 ± 12 years. Acute thrombi were observed in 224 (50%), and of these, ruptures were seen in 68% and erosions in 32%. Healed myocardial infarct was present in 40% of cases. Stable plaque in the absence of an acute thrombus in SCD patients was observed in 226 patients (50%), and of these organized thrombi were present in 97 (21%) and another 134 (29%) died with stable plaque in the absence of any acute or chronic total occlusion. Of the 134 patients with stable plaques, healed myocardial infarction in the absence of any thrombus was present in half the patients.
Thus in 69 patients the only cause of death was severe luminal narrowing (>75% cross-sectional area narrowing) in one or more coronary arteries (Figs. 1-9, 1-10 and 1-11). These stable plaques often show little necrotic core and are predominantly composed of collagen and calcified plaque. The incidence of healed plaque rupture in the stable plaques without healed myocardial infarction is 50%, which is much less than that seen in patients dying with acute plaque rupture, 75%. The highest incidence of healed plaque rupture is observed in patients dying with stable plaque and a healed myocardial infarction (80%).
Coronary Plaques in Acute Myocardial Infarction
The incidence of coronary thrombosis in hospital-based autopsies performed on patients with acute MI is >80%,39,40,41 with the exception of the study by Kragel et al., who report a lower 69% incidence of thrombosis.42 The reported incidence of thrombosis by angiography is as high as 90%. Separate studies by Davies et al. and Falk emphasized plaque rupture as the most important factor in the development of coronary thrombosis.40,43 However, Arbustini et al. reported in a series of 300 autopsies in patients with documented ECG proven and enzyme elevation that at least 25% of infarct-related coronary thrombi occurred from plaque erosion, and the incidence was
even higher among women.44 In our sudden death population at least 21% of patients showed evidence of an acute MI (AMI), and the incidence of thrombosis is 90%. Of these patients with AMI, thrombosis occurs in 70% of cases from plaque rupture and 30% from plaque erosion.26 In a series of studies by Davies et al., in a sudden coronary death population the incidence of AMI was 41%. Coronary thrombosis was found in 84% with reported chest pain within the week prior to AMI and death. Interestingly, in these AMI cases, 13 showed plaque fissure without a luminal thrombus. Another four of 32 cases (13%) studied without acute coronary events had myocardial infarction. Thus in Davies’ series, 13% of cases with AMI did not have any luminal thrombi. In a recent study by us of healed plaque rupture, AMIs were present in only 11% of sudden coronary deaths, whereas 80% had thrombi and 20% had no evidence of an acute event.37
even higher among women.44 In our sudden death population at least 21% of patients showed evidence of an acute MI (AMI), and the incidence of thrombosis is 90%. Of these patients with AMI, thrombosis occurs in 70% of cases from plaque rupture and 30% from plaque erosion.26 In a series of studies by Davies et al., in a sudden coronary death population the incidence of AMI was 41%. Coronary thrombosis was found in 84% with reported chest pain within the week prior to AMI and death. Interestingly, in these AMI cases, 13 showed plaque fissure without a luminal thrombus. Another four of 32 cases (13%) studied without acute coronary events had myocardial infarction. Thus in Davies’ series, 13% of cases with AMI did not have any luminal thrombi. In a recent study by us of healed plaque rupture, AMIs were present in only 11% of sudden coronary deaths, whereas 80% had thrombi and 20% had no evidence of an acute event.37
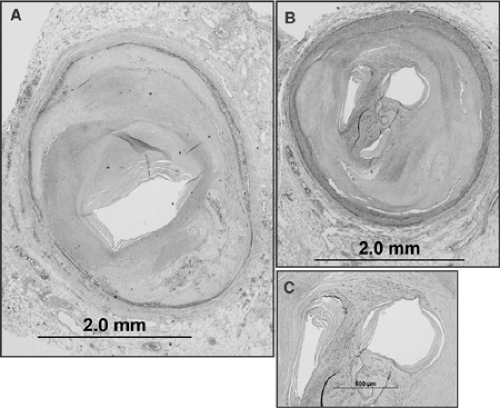 FIGURE 1-9. Histologic sections of stable atherosclerotic plaques with severe narrowing. A: The lesion shows a predominantly fibrous plaque with focal calcification and severe luminal narrowing. Note that in this stable lesion the media is extensively destroyed. B: A recanalyzed, fully organized thrombus with three centrally located large channels [(two of which are seen in (C) in high power)] with severe narrowing. Angiographically it would have appears as a single lumen with >90% diameter stenosis and not as a reanalyzed thrombus. |
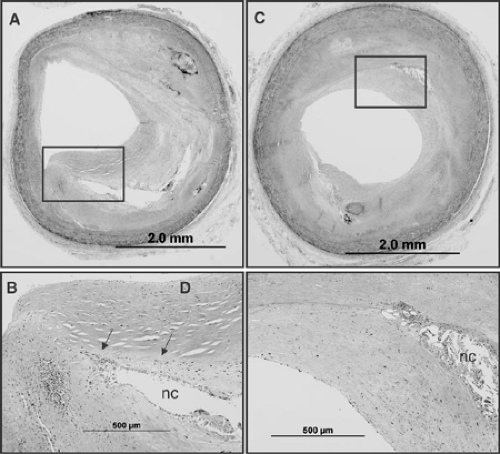 FIGURE 1-10. Photomicrographs of two adjacent sections (A) and (C) taken 4 mm apart, note presence of necrotic core (NC) with a recently organized healed plaque rupture. The boxed areas in (A) and (C) are shown at high power in (B) and (D). The area above the necrotic core consists of smooth muscle cells in a proteoglycan-rich matrix (area above NC outlined by arrows
Get Clinical Tree app for offline access
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|