Figure 14–1.
IKr, IKs, and Ito their respective genes and their role in the repolarization of the ventricular action potential
Pathologies due to mutation in ion channels were described clinically many years before an understanding of the underlying mechanisms. The Romano-ward and the Jervell and Lange-Nielsen syndromes, both congenital long QT syndromes, were first described clinically roughly 50 years ago [14, 15]. However, it wasn’t until 1995 that the link between K+ channels and action potential prolongation was made [16, 17]. As a result of these reports and the functional and structural knowledge of K+ channels, an understanding began of why certain drugs used to prevent cardiac arrhythmia could infact be pro-arrhythmic themselves [12 18]. Recently the same K+ channels implicated for Long QT have been found to be involved in familial atrial fibrillation (FAF) and the short QT syndrome(SQTS) [19, 20]. Further, mutation of Ito has now been implicated in the Brugada syndrome (BrS) Table 14.1.
Table 14–1.
K+ channel heritable arrhythmia genes and their associated currents and inherited diseases
Gene | KCNQ1 | KCNH2 | KCNE1 | KCNE2 | KCNE3 | KCND3 |
|---|---|---|---|---|---|---|
Function | α-subunit | α-subunit | β-subunit | β-subunit | β-subunit | α-subunit |
Current | IKs | IKr | IKs | IKr | Ito | |
Inherited disorder | LQT1 | LQT2 | LQTS | LQT6 | BrS | BrS |
FAF2 | FAF1 | |||||
SQT2 | SQT1 |
Voltage-Gated K+ Channels
The functional diversity among the primary voltage-gated K+ channels in the human heart can be classified in two broad classes. That of transient outward (Ito) and delayed-rectifying (IKr and IKs) K+ channels. Between atrial and ventricular tissues, and throughout the specialized conduction system these K+ channels are present at variable levels leading to a diversity in the morphology and duration of action potentials [21]. These different action potentials in various cardiac cell types are required to perform their specialized roles in the heart and typically are spatially arranged in a manner that is protective from arrhythmia. Dysfunction in the normal heterogeneity of action potentials brought on by K+ channel dysfunction is a potent pro-arrhythmic substrate.
Molecular Determinants of Voltage-Gated K+ Channels
Most of what is known about the biophysical basis of voltage-gated K+ channel gating is derived from studies of Shaker (Kv1.1) channels [22]. Voltage-gated K+ channels are typically composed of two primary subunits: The α-subunits are the pore-forming subunits that also contain a voltage sensing domain and a selectivity filter (see Fig. 14.2a). β-subunits are accessory subunits that affect the channel gating properties. In addition to these two canonical subunits, K+ channels can interact with a plethora of intracellular proteins including A-kinase anchoring proteins that can play a key role in their roles in cardiac physiology.
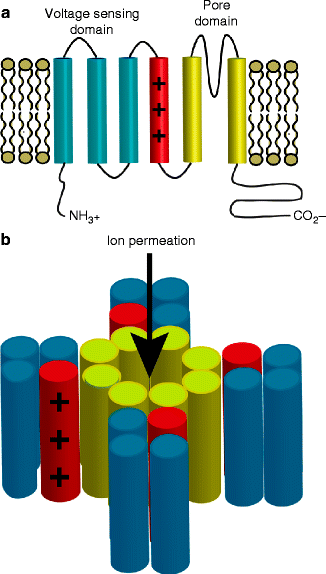
Figure 14–2.
Topology of voltage-gated K+ channels. (Panel a) α-subunit consist of six transmembrane helix with the first four largely responsible for voltage sensing and the last two forming part of the channel pore. (Panel b) Voltage-gated K+ channels are tetramers that assemble to make function channels with a central ion-conducting pore
α-subunits are composed of six transmembrane segments (S1 – S6) and functional voltage-gated K+ channels form by tetramerization of α-subunits with a central ion-conducting pore (see Fig. 14.2b). The first four transmembrane segments are primarily responsible for the channels response to voltage, with charged residues in S4 sensing changes in membrane potential [23, 24]. A conserved motif in the pore loop, between domains S5 and S6, of K + channels is responsible for ion selectivity.
The voltage-gated K+ channel accessory subunits are a diverse set of proteins. For IKr and IKs, they are largely modulated by the KCNE family of β-subunits that are small (∼100–150 amino acids) single transmembrane proteins. Another class of K+ channel subunits often co-assembling with faster opening and inactivating channels is typically larger intracellular proteins including KChaP, Kvβ1–3, and KChiP2.
Delayed Rectifying K+ Currents
Delayed-rectifying current was first described in sheep Purkinje fibers [25, 26]. Following those initial observations, characterization of the same current was made in atrial and ventricular cells as well as in pacemaker cells from various species [21].
The two prominent components first described in atrial and ventricular guinea pig myocytes based on their differences in time- and voltage-dependent properties were IKs (IK,slow) and IKr (IK,rapid) [27–29]. IKr is known to activate and inactivate rapidly and displays a marked inward rectification wherein ion conduction is favored inwardly. This rectification is a function of the channels recovering from inactivation faster than they deactivate. On the contrary, IKs does not display any inward rectification, minimally inactivates, and activates and deactivates slowly [28, 29]. These currents can be found in atrial and ventricular myocytes cells in most large mammals, including human, as well as in the purkinje fiber system [21]. In rodents, there are little if any delayed-rectifying channels as the AP is very brief and repolarized by rapidly opening K+ channels [30].
Congenital Channelopathies
In the following section, we will focus on congenital K+ channelopathies. Since the discovery that inherited QT prolongation could be caused by mutation in the K+ channel responsible for the rapidly and the slowly activating component of the delayed rectifier K+ current (IKr and IKs respectively), other congenital cardiac K+ channelopathies have been described, such as the short QT syndrome (SQTS, see Chap. 33), familial atrial fibrillation (AF, see Chap. 35), and the Brugada Syndrome (BrS, see Chap. 28).
Long QT Syndrome
The Long QT syndrome is a collection of cardiac disorders characterized by a prolongation of the QT interval on the ECG that affects an estimated 1 in 5,000–10,000 (see Fig. 14.3 and Chap. 27).
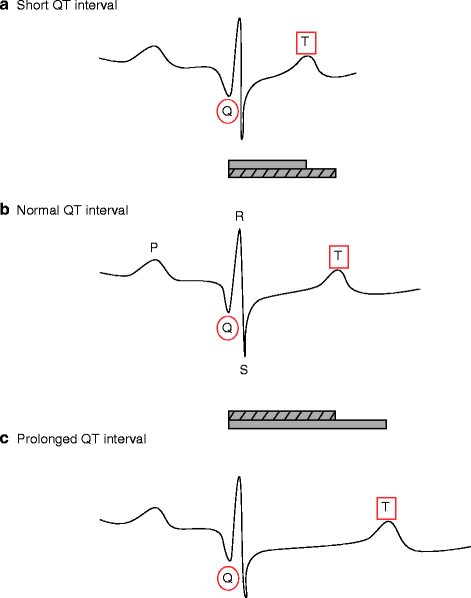
Figure 14–3.
Schematic representations of normal (b), short (a) and long (c) QT interval durations
The autosomal dominant inherited form of LQTS, originally called the Romano-Ward syndrome, is a collection of hundreds of mutations distributed over more than a dozen genes [31]. Modern nomenclature refers to these mutations as LQT1-LQT13. Another rare form of inherited LQTS is autosomal recessive and called the Jervell and Lange-Nielsen syndrome. In this syndrome, mutation in the genes responsible for IKs lead to cardiac arrhythmia and congenital deafness owing to these channels also being present in the inner ear [32]. To date five genes, KCNQ1, KCNE1, KCNH2, KCNE2, and AKAP9, that are directly responsible for the delayed rectifier K+ current (IKs) and the rapidly activating component of the delayed rectifier K+ current (IKr), have been implicated in LQTS.
IKs
IKs is a major contributor to cardiac action potential repolarization (phases 2 and 3, see Fig. 14.1). KCNQ1, like other voltage-gated K+ channels, forms a homotetramer to make function channels (see Fig. 14.2b) [17]. Expression of the alpha subunit, KCNQ1, alone in heterologous systems results in a K+ current that rapidly activates and partially inactivates(see Fig. 14.4a, 1) [33, 34]. The characteristic slowly activating, and slowly deactivating, current known as IKs is only observed with the coexpression of KCNE1 [33, 34]. At lower heart rates, IKs slowly activated over the course of an action potential and eventually leads to terminal repolarization. As rate increases and diastolic recovery times shorten, IKs fails to completely deactivate between beats and as a result a steady accumulation of open channels allows for critical rate-dependent shortening of the action potential [11]. This critical role of the channel allows for adequate diastolic filling time for the heart.
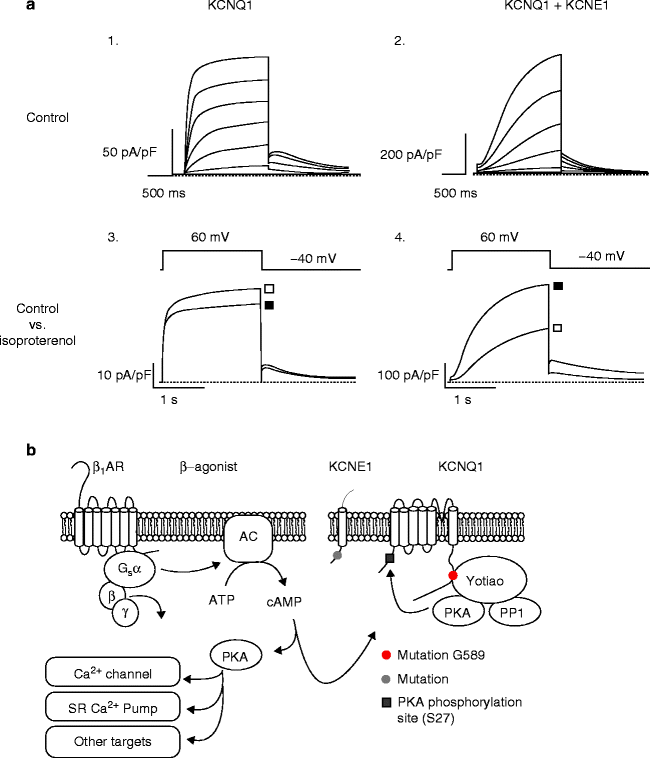
Figure 14–4.
(a) Currents recorded from KCNQ1 alone (A1) and when coexpressed with KCNE1 (A2) in Chinese hampster ovary cells. The effect of β-adrenergic stimulation on channel function (■) requires co-expression of both subunits, KCNQ1 and KCNE1 (A3 and A4), (□) control conditions. (b) Schematic of the β-adrenergic stimulation pathway, briefly binding of agonist to β-adrenergic receptors begins a cascade of actions that leads to protein kinase A (PKA) phosphorylating various cardiac targets, including KCNQ1 at serine 27 (S27)
Sympathetic Nervous Stimulation of IKs
Binding of epinephrine to G protein coupled β-adrenergic receptors (β-AR), leads to activation of adenylyl cyclase. Adenylyl cyclase subsequently synthesizes cyclic adenosine monophosphate (cAMP) from adenosine trisphosphate (ATP), which leads to an increased activity of the cAMP-dependent protein kinase A (PKA). In cardiac myocytes, PKA has a large number of downstream targets including IKs. Following β-AR stimulation, IKs amplitude increases, activation speeds, and deactivation slows (see Fig. 14.4). Interestingly, in heterologous systems, co-expression of KCNQ1 and KCNE1 alone is not capable of reproducing this behavior. The functional response of the channel to cAMP requires the presences of a third protein, A kinase anchoring protein 9 (AKAP9), also known as Yotiao [7]. AKAP9 tightly controls the posttranslational state of the channel by recruiting PKA, protein phosphatase 1, and phosphodiesterase PDED3. This highly evolved channel complex controlling the phosphorylation state is critical to the normal function of the heart, as changes in IKs function directly affect repolarization in the heart and its ability to respond appropriately to changes in rate.
IKs Related LQTs
Three variants of the Long QT syndrome are directly linked to defects in IKs function that lead to a prolonged cardiac action potential. These mutations decrease the total amount of K+ crossing the membrane during repolarization either by decreasing the number of channels on the cell surface, altering the channel’s voltage dependence and/or kinetics in such a way that IKs channel activity is reduced, or blunting the channel’s response to adrenergic stimulation.
Long QT syndrome variant 1 (LQT1) results from mutation of KCNQ1 and is the most common form of clinically observed inherited Long QT syndrome. Over 240 different mutations in KCNQ1 have been identified from patients presenting with Long QT. Long QT syndrome variant 5 (LQT5) results from mutation in KCNE1 and is a rarely occurring variant. More recently mutation in AKAP9 was found to underlie Long QT in a patient implicating a role for basal phosphorylation of the channel in cardiac myocytes [35].
IKr
The importance of hERG, standing for human ether-a-go-go-related gene, in normal human cardiac electrical activity became obvious when inherited mutation in KCNH2 was found to cause LQTS variant 2 [16, 36]. hERG (KCNH2) homotetramers form the channel pore and co-assemble with KCNE2 to generate cardiac IKr [37]. A physiological role of KCNE2 was elucidated when variants were identified as being associated with congenital and drug-induced LQT [31, 38, 39]. IKr kinetics are distinct from IKs in that despite both contributing to repolarization, they achieve this result via different mechanisms. For IKr, at depolarized potentials the channels open slowly and rapidly inactivate; however, during repolarization the channels recover from inactivation faster than they deactivate, leading to an increase in current critical to membrane repolarization in cardiac myocytes. IKr inactivation is a critical part of the channel function and is achieved by a C-type inactivation mechanism wherein a collapse of the selectivity filter reduces the ability of the channel to conduct K+ [12] (Fig. 14.5)
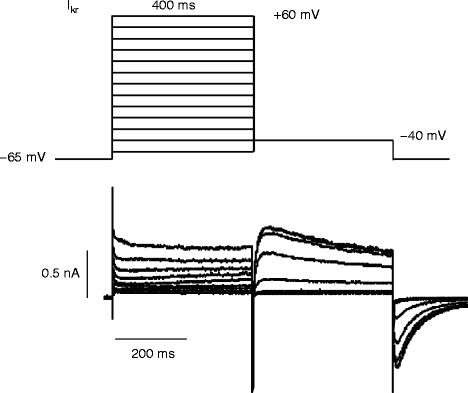
Figure 14–5.
The protocol used to record a current-voltage relationship of IKr and the resultant currents. Cells are first depolarized from a resting potential of −65 mV to different test potentials ranging from −60 mV to +60 mV for 400 ms and repolarized to −40 mV. Upon repolarization, recovery from inactivation procedes channel deactivation leading to larger currents
IKr Related LQTs
Like in IKs, mutation in both the α and β subunits of the IKr channel have been identified as causally linked to LQT2 and six respectively. LQT2 is the second most commonly observed LQT variant, behind LQT1, and is characterized by a reduction in IKr during repolarization that leads to prolongation of the action potential. This reduction in IKr is typically a result of trafficking defects that reduce the number of channels in the membrane but can also be the result of defects in activation, increased activation, or changes in ion conduction/selectivity [40, 41]. LQT6, which results from mutation in the β subunit KCNE2 also acts to reduce cardiac IKr and is much less commonly observed clinically.
Recently, small molecule activators of IKr have been identified that have the ability to increase current either through attenuation of inactivation or through a slowing of deactivation that can cause the channel to accumulate in the open state [42, 43]. This may have the potential to open a novel avenue for pharmacological intervention in some subclasses of LQTS.
Short QT Syndrome
Another rare inherited disease, the short QT syndrome (SQTS) [44], was identified in 2000 as coupled with paroxysmal AF. Further characterization showed that in one family, SQTS was linked to a history of ventricular fibrillation and sudden death [45].
Not surprisingly, for K+ channel SQTS, it was shown that mutations which increase K+ current led to the phenotype. Mutations have been identified in three different genes encoding subunits of K+ channels. The first mutations (SQT1) described were found in the gene KCNH2 where two different missense mutations, resulting in the same amino acid change (N588K), in the S5-P loop region of the cardiac IKr channel were identified. The mutation dramatically increases IKr, and reduces the affinity of the channel to IKr blockers [20]. Subsequently, a mutation has been found in a patient suffering from idiopathic ventricular fibrillation and a short QT interval. The mutation was a substitution of a valine by leucine at position 307 (V307L) in KCNQ1 (SQT2) and led to increased K+ current through IKs in a heterologous expression system [46]. SQT3 has most recently been observed as mutation in the gene KCNJ2 (D172N) that encodes for a K+ inward rectifier channel Kir2.1, the predominant component of cardiac IK1. Again, the phenotype is increased K + channel activity that leads to shorter APDs that underlie the clinically observed short QT interval. In SQTS, patients are noted to have a high incidence of arrhythmia, particularly atrial fibrillation [45, 47].
Atrial Fibrillation
Atrial fibrillation (AF) is a common cardiac arrhythmia, affecting millions in the US and leading to a twofold increase in mortality and a two to sixfold increase in stroke risk [48]. The vast majority of clinically observed AF is age-related. AF is characterized by a rapid and irregular atrial activation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


