, Domenico Corrado2 and Cristina Basso1
(1)
Cardiovascular Pathology Department of Cardiac, Thoracic and Vascular Sciences, University of Padua, Padova, Italy
(2)
Cardiology Department of Cardiac, Thoracic and Vascular Sciences, University of Padua, Padova, Italy
In nearly 15–20 % of SCDs in the young, a gross and histological examination of the heart fails to reveal a convincing pathological substrate which may explain the fatal outcome [1–4]. A thorough and complete autopsy, including the brain, results to be negative for an extracardiac or mechanical cardiac cause of death [5]. The heart is structurally normal and, by exclusion, the death is nothing but arrhythmic in origin. This death is known as “mors sine materia” and is frequently of familial occurrence, suggesting a hereditary genetically determined disorder [4]. Only the employment of molecular investigation may help to detect the genetic abnormality in the nucleotide DNA sequence, thus allowing to find that many SCDs sine materia are indeed ion channel diseases, now listed among nonstructural cardiomyopathies [6, 7]. Circumstances of death and the availability of the 12-lead ECG recorded during life are of utmost importance to address the investigation [5] (Fig. 9.1).
A leading cause of arrhythmic SCD without morphological substrate is long QT (LQT) syndrome, a genetically determined autosomal or recessive disease, characterized by a long QT interval (>450 msec) (Fig. 9.2) due to prolonged repolarization of the working myocardium, which causes an electrical vulnerability with possible onset of life-threatening arrhythmias, including torsades de pointes and ventricular fibrillation [8–11]. There are three main forms of LQT syndrome: LQT1 and LQT2 are mostly related to loss of function mutations in genes encoding potassium channel, whereas LQT3 is due to mutations of genes encoding sodium channel, namely SCN5A, the same gene involved in Brugada syndrome and Lenegre’s disease with AV block. Cardiac arrest occurs usually during exercise in LQT1, emotion in LQT2, and sleep in LQT3 [8].
The short QT syndrome, a Mendelian disorder with autosomal dominant pattern of inheritance, is much rarer than the LQT syndrome and is characterized by a QT interval shorter than 320 msec [12]. It has been linked to potassium gene mutations, with gain of function and shortening of the action potential. SCD occurs mostly during sleep.
Brugada (or Martini–Nava–Thiene) syndrome is an autosomal dominant inherited disorder characterized mostly by loss of function mutations of SCN5A sodium channel gene and featured by a nonischemic ST segment elevation at 12-lead ECG [13, 14]. Only 20 % of cases were found so far to have a genetic basis, whereas the remaining are still unexplained at molecular level [15, 16]. It is still a matter of debate whether hearts of patients dying suddenly with Brugada syndrome are structurally normal or present subtle changes (i.e., concealed structural cardiomyopathy), mimicking arrhythmogenic cardiomyopathy [13, 17, 18].
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a heredofamilial disorder of the calcium homeostasis [19, 20]. The defect involves the Ca++ uptake and release from the smooth sarcoplasmic reticulum, accounting for electromechanical coupling. The autosomal dominant form is characterized by a mutation of the ryanodine receptor 2 gene, which causes a Ca++ excessive release into the cytosol, impairing the sodium–calcium exchange at the level of the sarcolemma [20–23]. This interferes with myocyte repolarization and may trigger life-threatening arrhythmias. The autosomal recessive form is due to mutations of calsequestrin 2 gene, which regulates the reuptake of the calcium into the sarcoplasmic reticulum [24, 25].
The onset of life-threatening ventricular arrhythmias is rate dependent. When, during effort or emotion, the heart rate exceeds 120–125 bpm, onset of polymorphic ventricular arrhythmias and even ventricular fibrillation may occur. Stress test ECG is a fundamental tool to clinically unmask the disorder (Fig. 9.3) [26].
Many cases of SCD without substrate still remain unexplained, even after a thorough investigation of genes known to be related to the abovementioned syndromes. They still belong to the category of idiopathic ventricular fibrillation. Dysfunction of the Purkinje fibers has been advanced, with early repolarization wave at the ECG (the so-called Haissaguerre syndrome) [27].
With the genome-wide investigation technique, it is foreseeable that eventually a genetic explanation will be found in many other cases of “mors sine materia.” However, after the initial emphasis, caution is needed due to the difficult interpretation of the pathogenic significance of gene mutations in the absence of family history and cascade genetic screening [28–32].
9.1 Image Gallery
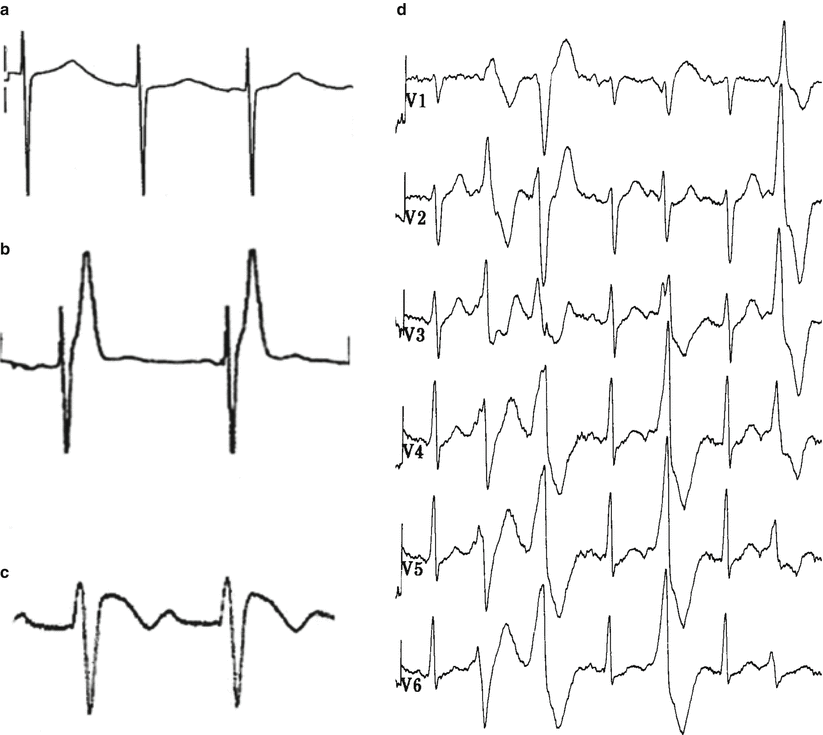
Fig. 9.1
ECG features in ion channel diseases at risk of sudden cardiac death. (a) Long QT syndrome. (b) Short QT syndrome. (c) Brugada syndrome. (d) Catecholaminergic polymorphic ventricular tachycardia (stress test ECG)
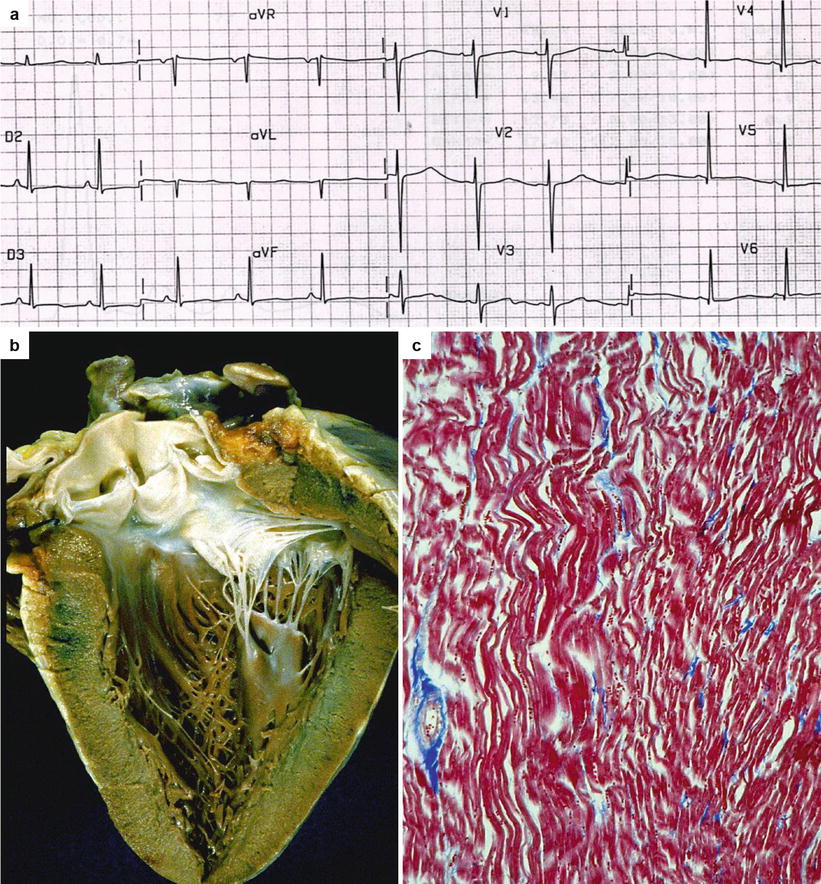
Fig. 9.2
Arrhythmic sudden cardiac death with normal heart in a 21-year-old young man while swimming. (a) 12-lead ECG showing the prolonged QT interval (QTc = 480 msec). (b) Grossly normal heart at autopsy. (c) Histology of the left ventricular myocardium reveals only waviness of the myofibers following cardiac arrest (Heidenhain trichrome)
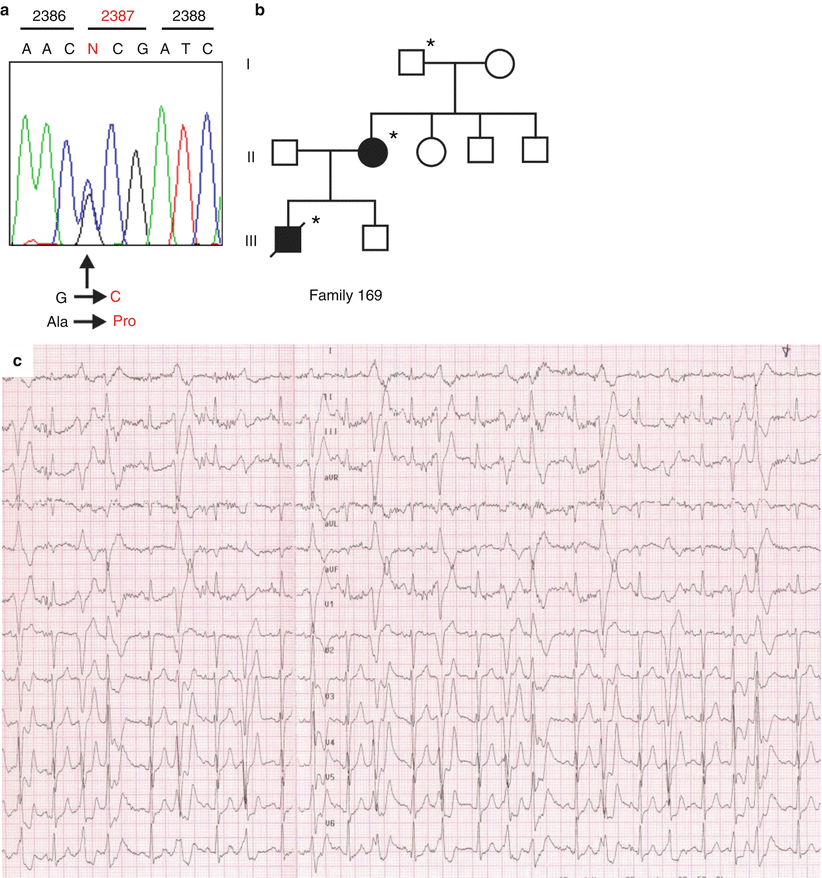
Fig. 9.3
Arrhythmic sudden cardiac death with normal heart in a 16-year-old boy on emotion. (a) Genetic analysis, performed at postmortem, of the DNA extracted from frozen spleen sample discovered a missense mutation (c.7160 G > C) at exon 47 of the ryanodine receptor 2 gene, accounting for amino acid change Ala2387Pro. (b) By extending the investigation to the family members, genetic screening revealed that the mother and the maternal grandfather carried the same mutation. (c) Polymorphous ventricular arrhythmias in the affected mother, triggered during stress test; (asterisc) indicates gene mutation carrier; black symbol indicates family member clinically affected by catecholaminergic polymorphic ventricular tachycardia
References
1.
2.
Thiene G. Sudden cardiac death and cardiovascular pathology: from anatomic theater to double helix. Am J Cardiol. 2014;114:1930–6.CrossRefPubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


