Usual interstitial pneumonia (UIP)
Bronchiolitis obliterans interstitial pneumonia (BIP)
Desquamative interstitial pneumonia (DIP)
Lymphoid interstitial pneumonia (LIP)
Giant cell interstitial pneumonia (GIP)
Later, Katzenstein and Askin refined this classification and added other entities such as nonspecific interstitial pneumonia (NSIP) and respiratory bronchiolitis-interstitial lung disease (RB-ILD). LIP and GIP were removed from the classification as they were considered no longer idiopathic diseases. BIP was also removed because it related to the acute lung injury pattern which eventually resolves and leaves no residual effects on the lung (Table 5.2).
Table 5.2
Current classification by Katzenstein, 1995
Usual interstitial pneumonia (UIP) |
Desquamative interstitial pneumonia (DIP) |
Acute interstitial pneumonia (AIP) |
Nonspecific interstitial pneumonia (NSIP) |
The most recent attempt in classifying these entities has been through the combined effort of the American Thoracic Society and the European Respiratory Society with two schemas in almost a decade [5–7].
As more knowledge accumulated, the classification became more refined, and criteria for diagnosis and subset of patients were recognized as different in prognosis and response to treatment. For example, patients with UIP associated with CVDs were found to represent a subset that fares better than the idiopathic UIP one [8]. The use of high-resolution CT and the experience of radiologist in diagnosing UIP made the requirement for surgical biopsy in many cases redundant.
Clinical and Radiological Findings of UIP
It is important before looking at the biopsy from interstitial lung disease to be familiar with the clinical and radiological findings. UIP usually presents in the fifth and sixth decades of life with an insidious onset of progressive dyspnea and shortness of breath. Patients have bilateral crackles on auscultation with a characteristic “Velcro type.” The pulmonary function tests (PFTs) have a restrictive pattern.
Radiologically, by high-resolution CT, there is a bilateral fibrosis accentuated in the lower portion of the lung and in the lung periphery with occasional honeycomb pattern in those areas. Ground-glass opacities are frequent findings in all of interstitial lung disease. The picture could be so characteristic that along with the clinical findings, in half the cases the diagnosis could be made without obtaining a biopsy.
Histopathologic Findings
Open lung biopsy used to be the gold standard for diagnosing idiopathic interstitial disease. In this biopsy several areas of the lung would be sampled preferably with a sample from each lobe targeting involved areas guided by the CT scan image. In the affected areas there is a patchy process of fibrosis and mild to moderate chronic inflammatory infiltrate. UIP could be called “peripheral fibrosis of the lung.” The process is usually more pronounced in the periphery of the lung with honeycomb changes in those areas and also in the periphery of the lung lobules with the center of the lobule less affected by fibrosis than the periphery. The fibrosis takes place by dense collagen deposition with a mild inflammation within those areas (Fig. 5.1). Fibroblastic foci are areas of recent fibrosis that show a recent type of fibrosis with myxoid background and small fascicles of fibroblasts within the interstitium juxtaposed immediately to areas of dense fibrosis, a feature that helps differentiating it from the fibroblastic plugs of cryptogenic organizing pneumonia in which the age of fibrosis is uniform and plugs assume a polypoid configuration (Fig. 5.2). The inflammation is never severe, but lymphoid aggregates could be present, and when there are several of them, a possibility of UIP associated with CVD should be raised to be confirmed by serological testing. The latter group of patients usually has a better prognosis than the one with idiopathic UIP [8]. Features that indicate chronic irritation of the lung could be seen in the same setting. These would include type II pneumocytes proliferation, bronchial metaplasia, and smooth muscle cirrhosis (Fig. 5.3). Honeycomb changes representative of those seen on CT are also present in the periphery under the pleura (Fig. 5.4). Foamy and pigmented macrophages could be seen in the alveolar spaces, but their significance and contribution to the diagnosis are limited. Rare granulomas or other incidental findings could be seen, but their significance and detraction from the diagnosis should be weighed against the other evidence of UIP.
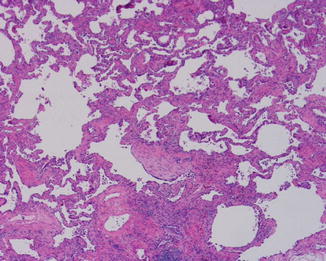
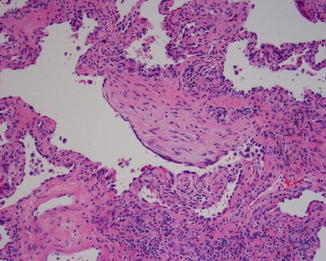
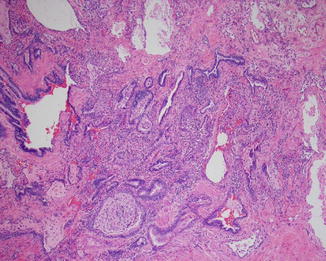
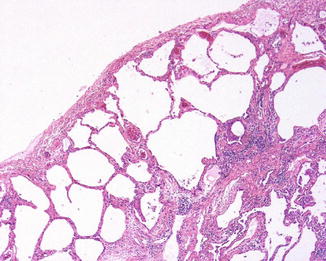
Fig. 5.1
Patch fibrosis with broad areas with collagen deposition alternating with areas with reticular fibrosis in UIP. In the center there is a fibroblastic focus
Fig. 5.2
The fibroblastic focus is under higher magnification showing the streaming fibroblasts in a myxoid background. The focus is only slightly bulging from the interstitium and is next to an area with dense fibrosis, in contrast to the polypoid fibroblastic plugs seen in BOOP
Fig. 5.3
An area of bronchial metaplasia with many alveolar spaces lined by columnar epithelium and inspissated mucous filling bronchial lumens
Fig. 5.4
An area with honeycomb changes under the pleural surface which corresponds to areas of accentuated fibrosis in the periphery of the lungs on high-resolution CT scan images
An important caveat to the diagnosis is the location of the biopsy as the tips of the lobes are notorious for harboring many of the features of chronic irritation of the lung with a sudden transition to the completely normal lung away from the very tip of the lobe. Caution should be exercised and correlation with high-resolution CT should be performed.
Recently, several reports of the possibility of making the diagnosis of UIP from transbronchial biopsy have surfaced [1]. However, the diagnostic yield of such biopsies is still low (specificity of 26–32 %) [3]. Another promising type of biopsy is the cryobiopsy which offers better size and less artifacts to make the diagnosis more reliable [3].
In the latest ATS/ERS schema for the diagnosis of UIP, the histopathologic findings were divided to definite, probable, possible, and not UIP based on the presence or absence of certain criteria summarized in Table 5.3.
Table 5.3
Revised American Thoracic Society/European Respiratory Society classification of idiopathic interstitial pneumonias: multidisciplinary diagnoses [7]
Major idiopathic interstitial pneumonias |
|---|
Idiopathic pulmonary fibrosis |
Idiopathic nonspecific interstitial pneumonia |
Respiratory bronchiolitis-interstitial lung disease |
Desquamative interstitial pneumonia |
Cryptogenic organizing pneumonia |
Acute interstitial pneumonia |
Rare idiopathic interstitial pneumonias |
Idiopathic lymphoid interstitial pneumonia |
Idiopathic pleuroparenchymal fibroelastosis |
Unclassifiable idiopathic interstitial pneumonias |
Clinical Course
Many UIP patients succumb to the condition known as “accelerated phase of UIP” where the patients go quickly into respiratory failure requiring mechanical ventilation. Histologically, the findings are similar to organizing diffuse alveolar damage superimposed or alternating with areas of characteristic UIP pattern. Hyaline membrane formation and fibroblastic proliferation along with edema and fibrin deposits are present along with interstitial dense fibrosis and evidence of chronic lung irritation.
Unfortunately, UIP presents a problem for understanding the underlying pathogenesis and also treatment. Immunosuppressive drugs failed to make any significant difference in the lives of these patients. Just recently, there are reports that pirfenidone, an anti-fibrogenic drug, shows some promising results in clinical trials [9]. Lung transplant is currently the only available hope for these patients.
Nonspecific Interstitial Pneumonia
It used to be reserved for non-classifiable cases that did not quite fit any of the idiopathic interstitial pneumonia category. However, in a landmark paper, Katzenstein et al. after analyzing a cohort of these cases reached to the conclusion that they represent a distinct group from UIP and other entities, even though they share similar features with UIP patients [10, 11].
Clinical and Radiological Findings of NSIP
These patients are usually a decade younger than those of UIP, and the male to female ratio is almost equal. They usually present with an insidious onset of dyspnea and shortness of breath. They have the same pattern of restrictive changes on the PFTs. Radiologically, there is a reticular pattern of fibrosis and ground-glass opacities on high-resolution CT. The characteristic peripheral attenuation seen in UIP is usually lacking, and honeycomb changes are not a standard feature. The picture is usually more prominent in the lower lobes though, which helps in the differential diagnosis with hypersensitivity pneumonia [12].
There is a frequent association with CVD, especially rheumatoid arthritis and scleroderma. NSIP could be the presenting disease in the lung of CVD. The prognosis of NSIP is much better than that of UIP with an overall 5-year survival between 75 and 90 % [11].
Histopathologic Features
The main distinguishing feature of NSIP from UIP, an important and crucial differential diagnosis, is the temporal and architectural uniformity of the fibrosis. There is a thickening of interalveolar septa by increased fibrosis and a chronic inflammatory infiltrate (Fig. 5.5). The amount of the latter could range from marked to sparse. Based on that, the NSIP patterns could be divided into cellular NSIP on one extreme and fibrotic NSIP with a possible third pattern of mixed NSIP when there are alternating patterns of both (Fig. 5.6). The fibrotic pattern imparts a worse prognosis and has been lumped with cases of UIP in the past. The key difference from UIP is the lack of fibroblastic foci and the lack of variegated pattern of fibrosis (Fig. 5.7). Microscopic foci of BOOP could be encountered in the biopsy, and these should not draw away from the diagnosis. The presence of granulomas or marked eosinophilia should point to other granulomatous diseases instead of NSIP.
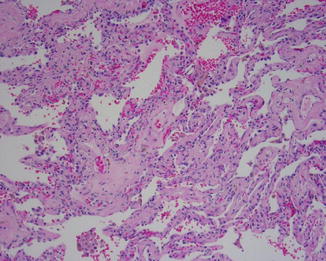
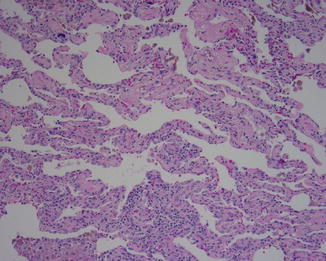
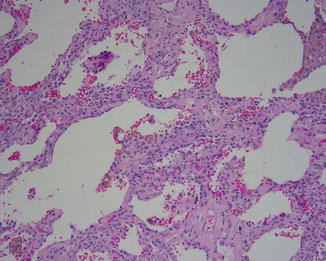
Fig. 5.5
Uniform fibrosis and moderate interstitial inflammation with uniform age of the fibrosis are all criteria for the diagnosis of nonspecific interstitial pneumonia (NSIP)
Fig. 5.6
NSIP showing the same type of uniform fibrosis and areas with markedly cellular infiltrate that lack the variegated pattern and fibroblastic foci seen in UIP
Fig. 5.7
NSIP with cellular interstitial chronic inflammation and uniform fibrosis
Clinical Course
While many clinicians wondered about the clinical entity of NSIP from the standpoint of clinical presentation, the fact remains that it is mostly a treatable disease and has a good response to corticosteroids, and it pays to distinguish it from other idiopathic interstitial pneumonias, especially UIP. As to what NSIP really represent, several speculations were put forward, including a possibility of CVD presenting in the lung with these manifestations in this group of patients, similar with what these diseases present in the kidney. Another possibility is that some cases could represent cases of hypersensitivity pneumonia where the insulting antigen has not been identified. Another possibilities are cases of slowly resolving BOOP or acute pneumonia and lastly cases of inadequately sampled UIP [13]. The entity is expanding, as will be discussed later, and the presence of macrophages, whether foamy or pigmented ones, should not detract from the diagnosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


