Fig. 9.1
Classification of interstitial lung disease (ILD). Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial pneumonia (IIP) and is the archetypal fibrotic lung disease. All ILDs however can result in progressive fibrotic lung disease, worsening symptoms, impaired gas exchange, and ultimately death (The figure describes a classification of ILD adapted from the 2002 international consensus [3]).CTDconnective tissue disease,HPhypersensitivity pneumonitis,LAMlymphangioleiomyomatosis,LCHLangerhans cell histiocytosis,DIPdesquamative interstitial pneumonia,NSIPnon specific interstitial pneumonia,LIPlymphoid interstitial pneumonia,RBILDrespiratory bronchiolitis ILD,AIPacute interstitial pneumonia,COPcryptogenic organizing pneumonia
Many forms of ILD are idiopathic, where no etiological factors can be identified, whereas others are clearly linked to connective tissue diseases, prescribed drugs, cigarette smoking, and environmental exposures to antigens or dusts. The most common ILDs are idiopathic pulmonary fibrosis (IPF) and other idiopathic interstitial pneumonias (IIPs), sarcoidosis, connective tissue disease-associated ILD, and hypersensitivity pneumonitis (also known as extrinsic allergic alveolitis).
Interstitial lung diseases commonly present with dry cough and breathlessness. Examination findings may confirm finger clubbing and crackles on auscultation whilst chest radiography may show diffuse abnormalities including infiltrates, fibrosis, and reduced lung volumes. The loss of lung volume typical of ILD is usually confirmed with restrictive lung function tests and reduced gas-transfer measurements. Often there is a delay in diagnosis whilst patients are treated for lower respiratory tract infections or for pulmonary edema, conditions more prevalent than ILD that commonly form a differential diagnosis until a full clinical assessment including definitive imaging with high-resolution computed tomography (HRCT) is performed (Fig.9.2).
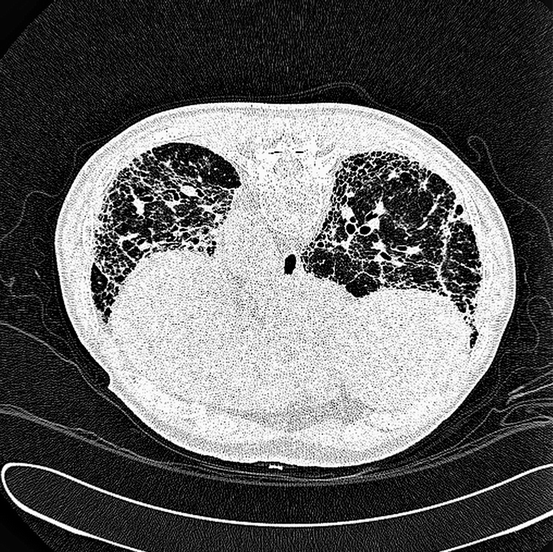
Fig. 9.2
Imaging of IPF. High-resolution CT scan (HRCT) of an 82-year-old male patient with advanced IPF. The image, acquired prone, shows evidence of severe basal fibrosis in a typical sub-pleural distribution, minimal “ground-glass” change, “honeycomb” lung, and evidence of secondary bronchial dilatation. The patient had presented with progressive breathlessness and dry cough. Lung function tests confirmed severe intrapulmonary restriction with low lung volumes and impaired gas transfer. The patient developed worsening respiratory failure and died 18 months from presentation
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is the most common ILD and is the archetypal progressive fibrotic lung disease. IPF is a specific form of chronic fibrosing interstitial lung disease of unknown cause that has an estimated incidence of 5–10 per 100,000 population/year, with the incidence generally accepted to be rising. It is believed that over 500,000 patients are affected by IPF in the USA and Europe. IPF is typically a disease of the elderly, with a mean age of onset of 67 years and is more common in smokers. IPF is characterized by relentless progression of lung fibrosis, impaired lung function, worsening gas exchange, and prominent symptoms of breathlessness and dry cough. Survival of patients with IPF is worse than for many cancers with a median survival of only 3 years from diagnosis.
Whilst population survival in IPF is predictably poor, it is recognized that there is heterogeneity in the clinical course of the disease as well as unpredictable acute worsening or exacerbations of IPF [4]. There appear to be several possible natural histories for patients with IPF which makes accurate prognostication difficult and planning for palliative care and end of life care a particular challenge. The possible natural histories for IPF are summarized in Fig.9.3. A practical and accurate method to predict the course of the disease and the survival in individual patients has been proposed [5]. In this study, the 1 year mortality of patients with IPF appeared to be predictable, based on modeling retrospective clinical trial data of over 1,000 patients with IPF, using four clinical parameters; age, respiratory hospitalization, forced vital capacity (FVC), expressed as a percentage of the predicted value, and 24 week change in FVC. This approach may facilitate decision- making by patients, carers, and clinical teams, although it clearly needs careful prospective evaluation and validation in other IPF populations before firm recommendations regarding prognostication and guiding clinical management can be made.
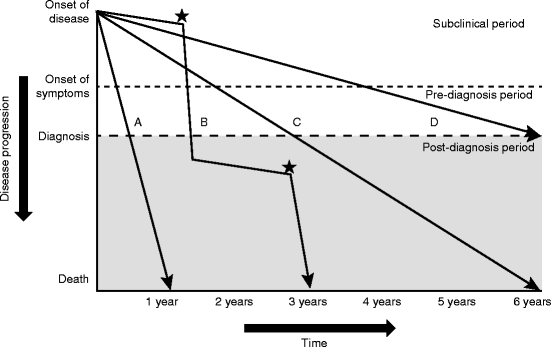
Fig. 9.3
The natural history of IPF (Reproduced with permission from Ley et al. [4]). The variable and unpredictable clinical course of IPF is represented schematically. Following the onset of disease which may be only detectable on high-resolution CT scan or chest radiograph there is a subclinical period. There follows a symptomatic period which is typically present for some time before the diagnosis is secured. The rate of decline may be rapid (A), more slowly progressive (C) and (D) or mixed (B) where a relatively indolent deterioration is punctuated with periods of acute decline (*) due to recognized complications such as pneumonia or unexplained AE-IPF. These deteriorations may be fatal or leave the patient with significantly worsened disease
A number of patients are recognized to have acute worsening of their disease due to identifiable conditions such as pneumonia, pneumothorax, pulmonary embolism, acute coronary syndromes, or cardiac failure. However, it has been estimated that up to 15 % of patients will experience an acute exacerbation of IPF (AE-IPF) each year [6]. These exacerbations are characterized by an acute deterioration (<30 days), with new or worsening breathlessness often accompanied by cough, fever, and flu-like symptoms. Imaging confirms new bilateral ground glass shadowing whilst no other explanation for the exacerbation, including infection, can be identified. Whilst the mechanism of AE-IPF remains unclear and may simply represent an acute acceleration of the fibrotic process underpinning IPF, some authors have speculated that occult viral infection or gastro-esophageal reflux disease may play a role [7]. There is no strong evidence on which to base recommendations for the optimal treatment of AE-IPF, although recent guidelines recommend the use of corticosteroids in the majority of these patients [8]. These exacerbations can have a serious impact on both the quality of life and survival of patients with IPF, with up to 50 % of patients dying during an exacerbation [6].
IPF is associated with other diseases that can contribute to worsening symptoms, deterioration and death. It has been recognized that patients with IPF are at significantly increased risk of lung cancer, acute coronary syndromes, and pulmonary embolism. In addition, comorbidities such as pulmonary hypertension and chronic obstructive pulmonary disease (COPD) may contribute to the disease course whilst treatments such as radiotherapy, surgery, and potentially chemotherapy for co-existent lung cancer can precipitate a deterioration or AE-IPF [8,9].
Intensive Care for Patients with ILD?
Patients with IPF in whom the disease progresses to a stage of respiratory failure may trigger a referral to the intensive care unit (ICU) for consideration of ventilatory support. Available data however show that the outcome for these patients is very poor, mechanical ventilation is mostly futile, and ICU support for these patients is usually not appropriate due to mortality rates of up to 100 % [1]. It is important for the clinical team to be aware of the clinical course of the patient’s disease. If a patient has demonstrated the typical inexorable decline in lung function, gas exchange, and symptoms that characterize IPF, it is not appropriate to offer ICU-based organ support. More challenging is the patient with IPF who appears to have an acute deterioration or AE-IPF. In this situation, ICU may be an appropriate setting to perform in a safe and timely fashion the necessary extended investigation to exclude any reversible cause of deterioration in these patients [10]. Patients and their families should be informed about the prognosis, outcome, and overall outlook before making decisions about ventilation and organ support. It is important that all clinicians involved in the management of ILD are aware of the issues relating to the ICU care of these patients and ideally have agreed institutional guidelines which the medical, ICU, and palliative care teams can share [11].
Treatment of IPF
Lee et al. propose a “3 pillars of care” model of for patients with IPF which focuses on (1) disease-centered management, (2) symptom-centered management, and (3) education and self-management [12].
Disease-Centered Management
It is clear that pharmacological treatments aimed at reversing the progressive fibrosis that characterizes IPF have been unsuccessful and often harmful because of significant adverse effects. Traditionally steroids have been used to treat IPF, although no clinical trial data to support their use exist. In the British Thoracic Society ILD Guidelines 2008, a recommendation to use triple therapy with prednisolone, azathioprine, and n-acetylcysteine was made [1]. However, subsequent international guidelines have been unable to recommend the routine use of immunosuppressive drugs and indeed concluded that there is no proven pharmacological therapy for IPF [8]. A recent randomized controlled trial of triple therapy (PANTHER-IPF) was stopped prematurely as patients treated with triple therapy had increased mortality, more serious adverse events, and drug discontinuations, without evidence of benefit. Many other disease-modifying therapies have been tried unsuccessfully and whilst there may be some benefit from the anti-fibrotic drug pirfenidone in selected patients, there is unfortunately no effective drug treatment for progressive IPF. Patients with IPF should be offered participation in well-designed clinical trials with the potential benefits it may bring to both them and other patients in terms of outcomes, symptoms, and quality of life made explicit, although countered by the potential for adverse effects and harm [8].
The use of long-term oxygen therapy (LTOT) has been recommended for patients with IPF who have resting hypoxemia using the same criteria as recommended for patients with COPD [1,8]. It remains unclear however in the absence of clinical trial data that the survival benefits of LTOT seen in patients with COPD can be extrapolated to patients with IPF.
Lung transplantation is an effective treatment for a minority of patients with IPF. In selected patients, transplantation can dramatically improve the disease trajectory, although timing of referral for assessment remains critical to prevent patients becoming too unwell for consideration of lung transplant or dying on the waiting list [13]. Unfortunately many patients with IPF are elderly and have comorbidities which make them unsuitable for transplantation.
Symptom-Centered Management
The focus on symptom-centered management links with the concept of “best supportive care” as outlined in the British Thoracic Society guidelines and is clearly integral to effective palliative care for a disease which has no proven cure or disease-modifying treatment [1,8,12].
An important consideration in being able to proactively identify patients with progressive disease and worsening symptoms is careful monitoring using appropriate clinical evaluation, symptom scores, and quality of life assessments. This approach allows the clinical team to consider therapy choices but also helps patients understand their disease course. The standard objective measures of disease severity and progression are lung function as measured by forced vital capacity (FVC), gas transfer (TLco), oxygenation saturation, and 6 min walk distance. It is widely accepted that a fall in FVC of ≥10 % or TLco of ≥15 % over a 3–6 month period is indicative of disease progression and a poor outcome. It is clearly important however to ensure that the patient’s symptoms are assessed and quantified accurately. Breathlessness is almost universal in patients with IPF, although there is a paucity of data on the clinical utility of assessment tools, most of which were developed for assessment of COPD patients, in this group of patients. Breathlessness has been demonstrated to have a strong correlation with both quality of life and mortality in IPF [14]. Despite its prevalence and prominence, breathlessness has not been the focus of many studies of the natural history or treatment interventions in IPF. The use of a patient-reported measure of breathlessness, Dyspnea-12, has shown promise as a simple, reliable, and valid instrument in patients with ILD [15].
Whilst breathlessness in IPF is principally due to the physiological impact of fibrosis, reduced gas exchange, and ventilation-perfusion mismatching, other factors also influence the patient’s perception of breathlessness. In a cross-sectional study of patients with ILD some of whom had IPF, both depression and functional status, as measured by walk distance, were strongly associated with breathlessness [16]. The authors of this study speculated that treatment of depression and programmes of exercise may improve both breathlessness and quality of life.
A dry irritating cough can be a distressing symptom for IPF patients [17]. Cough in IPF is often refractory to anti-tussive therapies and it may be a predictor of progressive disease and mortality [18]. The mechanism of cough in IPF is poorly understood, although it may be due to heightened cough reflex sensitivity [19]. Other co-existent causes of cough, such as gastro-esophageal reflux, upper airway cough syndrome, asthma, and COPD should be considered. There is no IPF-specific tool to assess the severity or impact of a patient’s cough, although there appears to be a strong correlation between cough frequency and cough-related quality of life measures [20].
Fatigue in IPF can be a dominant symptom for some patients and can be exacerbated further by muscle deconditioning [17]. Many patients with IPF suffer from anxiety and depression, which can be compounded by the effects of chronic and debilitating symptoms, adverse effects of drug therapies, and fear of disease progression and death [16].
Health-related quality of life (HRQOL) in IPF is now recognized as an important measure to identify changes in the disease process and to quantify the impact of symptoms in individual patients. It is also being increasingly used as an outcome measure in clinical trials. Swigris et al., having recognized the lack of validated HRQOL tools in this disease, interviewed patients in order to identify themes and develop an IPF-specific tool ATAQ-IPF [17,21]. A modification (SQRQ-I) to the Saint George’s Respiratory Questionnaire has been proposed to be reliable for measuring health-related quality of life in patients with IPF [22]. More recently, the King’s brief interstitial lung disease questionnaire (K-BILD) has been proposed as a simple tool to assess HRQL in three domains of psychological impact, breathlessness/activities, and chest symptoms [23]. In a pilot study, HRQOL was found to be significantly lower in patients with IPF compared to other ILDs and was also lower in patients with IPF who were prescribed long-term oxygen therapy or immunosuppressant medication. Clearly more studies are needed to assess the longitudinal changes in HRQL using IPF-specific tools and the potential impact of interventions both in research trials and in clinical practice.
Specific Symptom-Centered Treatment
There are a number of approaches that form the foundation of “best supportive care” for patients with IPF. These include smoking cessation, addressing nutrition, treating right heart failure and influenza and pneumococcal vaccination that are likely to improve symptoms and prevent the development of complications. In addition, there are four principal symptoms that should be sought and considered in all patients with IPF.
Breathlessness
A recent systematic review concluded that there were scant data, too much trial heterogeneity to facilitate meta-analysis, and little robust evidence to support the use of specific treatments for the relief of breathlessness in IPF patients [24]. The review suggested that supplemental oxygen, pulmonary rehabilitation, and opioids may be beneficial. BTS guidelines recognize that in the absence of suitable controlled studies of long-term, short burst or ambulatory oxygen therapy in ILD, recommendations are extrapolated from studies in COPD. The guidelines recommend that patients with persistent resting hypoxemia should be considered for palliative oxygen at home delivered by an oxygen concentrator. These individuals may also benefit from ambulatory oxygen if they remain active outside the home. Patients who are not chronically hypoxic but who are breathless, mobile, and exhibit desaturation on exercise should be considered for ambulatory oxygen if improvement in exercise capacity or less breathlessness can be demonstrated by formal ambulatory oxygen assessment. Intermittent supplemental oxygen for periods of 10–20 min at a time and delivered by oxygen cylinder (short burst oxygen therapy) may relieve breathlessness associated with hypoxemia in patients with IPF who do not require an oxygen concentrator or ambulatory oxygen.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


