Fig. 11.1
Growth factor signaling between epicardium, endocardium, and myocardium during midgestation. Details are described in the text
11.2.2 Myocardial-Endocardial Signaling
The myocardium plays a strong paracrine and autocrine role in the regulation of cardiac development and growth and has an important impact on the endocardium as well as on endothelial cells (e.g., from capillaries). It was demonstrated, for example, that cardiomyocytes are the main source of vascular endothelial growth factor A (VEGF-A), which is one of the most potent angiogenesis-inducing factors of the body [8]. Cardiomyocyte-specific VEGF-A knockout reduces the level of VEGF-A mRNA to <15 % of the wild-type level and leads to significant embryonic mortality and hypovascular, thin-walled hearts [8]. VEGF-A mainly binds and activates the receptor tyrosine kinase VEGF receptor2 (VEGFR2, also called Flk1), which is the major mediator of VEGF-A-dependent mitogenic, angiogenic, and permeability-enhancing effects. VEGFR2 is expressed in endothelial cells including the endocardium. VEGFR2 knockout mice die at embryonic day E8.5 – E9.5 and completely lack the endocardium, blood vessels, and cardiac trabeculation [9]. Thus, myocardial-derived VEGF-A is functionally important for the formation of the endocardium, which in turn provides crucial signals for myocardial development (Fig. 11.1). In a similar manner, cardiomyocyte-derived angiopoietin-1 binds to its receptor Tie2 (expressed on endothelial and endocardial cells) to induce the formation of the endocardium [3].
An important area of signaling between the endocardium and myocardium is found in the development of endocardial cushions in the outflow tract (OFT) and the atrioventricular canal (AVC) that later form the cardiac valves [10]. The endocardial cushions are expansions of the cardiac jelly and become visible at embryonic day E9 of mouse development. Endocardial cells lining these cushions become mesenchymal cells and move into the cushions to form the future valve interstitial cells. This process is called epithelial-to-mesenchymal transition (EMT) and is triggered by myocardial signals, for example, by bone morphogenic protein (BMP)2, which is expressed first in the myocardium surrounding the AVC [10]. Its importance for endocardial EMT is shown in cardiomyocyte-specific BMP2 deleted mice, which exerts disturbed AV cushion formation [11]. BMPs bind to type I (in the myocardium ALK2/3) and type II receptors (in the myocardium encoded by Bmpr2) at the surface of its target cell and trigger SMAD1/5/8-dependent signaling. Endocardial-specific ablation of Alk2, Alk3, or Bmp2r results in strongly impaired EMT [10]. Cardiomyocyte-derived transforming growth factor (TGF) β2 and VEGF-A are similarly involved in the regulation of endocardial EMT, whereby it has been suggested that precise levels of VEGF-A are essential for this process, since both overexpression and inhibition of VEGF-A lead to impaired EMT [10]. In this regard, deregulated myocardial VEGF-A levels were proposed to account for endocardial cushion defects due to environmental stresses or Down syndrome. Some cardiomyocyte derived growth factors also act in an autocrine fashion to regulate formation of the ventricular myocardium. Cardiomyocyte-derived BMP10, for instance, induces cardiomyocyte proliferation and maintains a normal expression level of key cardiogenic factors like NK2 homeobox 5 (NKX2-5) and myocyte enhancer factor 2c (MEF2c); lack of BMP10 in mice results in embryonic mortality between E9.5 and E10.5 with a hypoplastic myocardium [12].
11.2.3 Epicardial-Myocardial Signaling
During midgestation, around E10.5, the epicardium is beginning to form as the outmost epithelial layer that ensheathes the heart in parallel to growth of the myocardium and formation of the coronary vasculature [10]. Before that time point, the linear and looping heart tube consists only of two layers, the outer myocardium and an inner layer of endothelial cells (endocardium). The developing epicardium promotes cardiomyocyte proliferation and thereby the proper development of the myocardial compact layer, which is extremely thin after experimental removal of the epicardium in chick embryos. This regulatory role is mainly mediated by the release of paracrine factors: FGF 9, FGF16, and FGF20 are produced in the epicardium and act on FGFR1/FGFR2 on cardiomyocytes in the adjacent compact layer to induce cellular proliferation and modulate differentiation (Fig. 11.1) [3]. FGF effects in the myocardium also trigger activation of hedgehog (HH) signaling [13]; this circuit is comprised of three ligands, sonic (S)HH, indian (I)HH, and desert D(HH), which are all expressed in the embryonic and adult heart, SHH being the most abundant. At midgestation, SHH is expressed in the epicardium, but not in the myocardium. HH ligands bind to the cell surface receptor patched (PTC). In the absence of ligand, PTC inhibits the activation of the multi-transmembrane protein Smoothend (Smo). Upon HH ligand binding, Smo triggers downstream signaling that culminates in activation of GLI transcription factors. Epicardial-derived SHH binds to PTC, which is expressed on cardiomyoblasts and perivascular mesenchymal cells in the myocardium and induces the expression and release of multiple proangiogenic molecules, especially VEGF-A, VEGF-B, VEGF-C, and angiopoietin2 from its target cells. These factors, in turn, promote coronary vascular development, which starts in the subepicardial space with the formation of the primary vascular plexus (between E11.5 and E13.5) that is later remodeled and expands into the myocardium to form the mature coronary tree. Inhibition of HH signaling in whole heart cultures with the specific inhibitor cyclopamine completely blunts coronary plexus formation, while transgenic overexpression of the HH transcription factor GLI2 in the myocardium at E13.5 increased the vessel density in the subepicardium [14]. Interestingly, HH signaling is still important for maintenance of the coronary vasculature in adult mice, as induced genetic deletion of PTC in cardiomyocytes leads to diminution of cardiac capillarization, aggravation of cardiac hypoxia, cardiomyocyte cell death, subsequent heart failure, and death. Inhibition of HH signaling after myocardial infarction (MI) triggers increased scarring, heart failure, and lethality. Of note, the ligand SHH is expressed in perivascular and interstitial fibroblasts in the myocardium of adult mice, which are cells that are originally mainly derived from the epicardium. In addition, the epicardium itself becomes reactivated and expands after MI and starts to release paracrine factors like WNT1, VEGF-A, FGF, TGFβ2, stromal cell-derived factor 1 (SDF1), and monocyte chemoattractant protein 1 (MCP1) [15, 16]. These factors play a protective role when administered to the myocardium after MI [16].
11.2.4 Myocardial-Epicardial Signaling
During embryonic development, the epicardium does not only play a prominent regulatory role via the release of paracrine factors but also contributes to different cell types (mainly fibroblasts and vascular smooth muscle cells) in the heart through EMT. Epicardial cells start to undergo EMT (starting at around E11.5 in mice) to form mesenchymal cells known as epicardium-derived cells (EPDCs, Fig. 11.1) [10, 13]. EPDCs reside in the subepicardial space and also migrate into the myocardium to form a subset of cardiac fibroblasts and vascular smooth muscle cells. Although previously it was thought that all capillary endothelial cells also arise from EPDCs, recent data revealed that only a small fraction of these cells originate from the epicardium [13]. The majority of vascular endothelial cells are derived from the endocardium and the sinus venosus. Epicardial cell proliferation and activation as well as EMT occur during myocardial injury such as MI also in adult mice. This leads to a thickened epicardial layer on the cardiac surface, but in contrast to embryonic development, not to an invasion of the subjacent myocardium. Epicardial EMT mainly is triggered by myocardial-derived growth factors. In this regard, Wnt ligands (WNTs) from the myocardium (e.g., WNT8a or WNT9) but also autocrine epicardial WNT1 act on the epicardium [3]. In their canonical signaling mode, WNTs bind to a co-receptor complex consisting of frizzled (Fzd) family seven-pass transmembrane proteins and the lipoprotein receptor related 5/6 (LRP5/6), which ultimately leads to stabilization and nuclear translocation of β-catenin. β-Catenin forms a complex with LEF/TCF family DNA binding proteins to activate the transcription of WNT target genes. Elimination of β-catenin from the epicardium with a Gata5-Cre leads to embryonic mortality in mice between embryonic day 15 and birth, whereby the mutant mice display a defective coronary artery development, but normal veins and microvasculature. This phenotype is the consequence of failed expansion of the subepicardial space and impaired differentiation of epicardium-derived coronary smooth muscle cells, indicating a defective EMT in response to ablation of WNT signaling. In a similar manner, FGF10 from the myocardium might act on FGF receptors-1/2 on epicardial cells. Elimination of this signaling axis leads to myocardial hypoplasia and a defect in cardiac fibroblast formation, although this was disputed in one study [17, 18]. In a third important pathway, platelet-derived growth factor (PDGF) signaling from the myocardium regulates epicardial EMT. When both epicardial PDGF receptors α and β are eliminated from epicardial cells, the process of EMT is blocked with a complete lack of EPDCs. However, as revealed by a selective knockout of the α- and β- form of the receptor in the epicardium, both receptors also have independent functions: the PDGF receptor α promotes the development of myocardial fibroblasts, while the PDGF receptor β promotes the formation of coronary vascular smooth muscle cells.
11.3 Intercellular Signaling During Pathological Cardiac Growth
While intercellular coordination during the embryonic phase, when the developing heart is still small, depends mainly on signals from the epicardium and endocardium, in the adult heart – due to larger distances – interspersed cells such as fibroblasts and capillary endothelial cells become a more important source of regulatory growth factors that impact cardiac myocytes.
11.3.1 Communication Between Cardiomyocytes
Although in terms of cell number cardiomyocytes represent only 30–45 % (depending on species) of all heart cells, because of their size they contribute to more than 90 % of the heart’s volume. Rich communication takes place among cardiomyocytes. Importantly, cardiac myocytes are directly coupled via gap junction at the intercalated disk (Fig. 11.2) [19]. Mainly ions (Ca++) and small solutes pass through gap junction to promote impulse conduction in the cardiac conduction system and the working myocardium. The gap junctions in adult mice are constituted by connexin40 in myocytes of the conduction system, but by connexin43 in the working myocardium. Besides impulse conduction, the connexins are important for cardiac morphogenesis, since heterozygous and homozygous deletion of connexin40 leads to developmental abnormalities of the heart, including double-outlet right ventricle, tetralogy of Fallot, and endocardial cushion defects [20]. Cardiomyocyte-specific deletion of connexin43 results in outflow tract abnormalities and cardiac hypertrophy after birth.
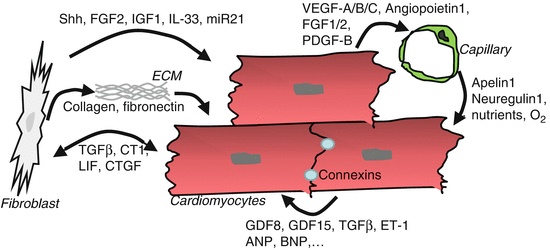
Fig. 11.2
Intercellular communication in the adult myocardium as it occurs during hemodynamic overload. Details are described in the text
Cardiomyocytes also secrete various growth factors, which either act in an autocrine fashion on the secreting cell, a neighboring cardiomyocyte or on non-cardiomyocytes. Among the myocyte-derived factors with functional autocrine effects are, for example, endothelin 1, ANP, and BNP as well as multiple TGFβ family members, including TGFβ, growth differentiation factor (GDF) 15, and myostatin (GDF8). TGFβ is released from cardiac fibroblasts, but also from cardiomyocytes. Ablation of the TGFβ receptor (R)2 specifically in cardiac myocytes strongly reduced cardiac hypertrophy, fibrosis, and improved cardiac function during experimental pressure overload in mice by transverse aortic constriction (TAC). In this procedure, which often is used as a model of human disease in mice, a ligature is placed between the left common carotid artery and the right innominate artery around the ascending aortic arch. This leads to a robust mechanical pressure overloading of the left ventricle with cardiac hypertrophy (+30–60 % in heart weight) within 2 weeks and reduced cardiac function within 4–8 weeks after surgery. The data from cardiomyocyte-specific Tgfβr2 knockout mice indicate that intrinsic cardiomyocyte TGFβ signaling is a strong promoter of pathological hypertrophy and dysfunction under these circumstances. In contrast, cardiomyocyte GDF15 acts to inhibit hypertrophy and death in these cells. Myocardial myostatin is crucial for the maintenance of cardiac homeostasis, as its selective-induced genetic ablation under baseline conditions resulted in hypertrophy, heart failure, and death associated with metabolic imbalance and over-activation of the AMP-activated kinase (AMPK) [21].
11.3.2 Endothelial-Cardiomyocyte Crosstalk
As an organ highly dependent on oxidative energy production, the capillary density in the heart is high, and each cardiomyocyte is supplied roughly by one capillary [22]. Capillary endothelial cells are closely associated with cardiomyocytes in an ideal diffusion range for capillary-derived nutrients and oxygen but also for reciprocal paracrine signals between these cells. It has been demonstrated that cardiomyocytes regulate the formation and adaptation of the myocardial capillary network and that angiogenesis (i.e., the formation of capillaries from preexisting endothelial cells) is enhanced during increased hemodynamic load and cardiac hypertrophy in multiple different species (mice, sheep, humans). In fact, this increase in myocardial capillaries (by about 30–50 %) is important for the preservation of cardiac function during hypertrophy. How is myocardial angiogenesis regulated, especially during pathological overload? As stated above, around 85 % of the VEGF-A within the heart is produced by cardiac myocytes [8]. Similarly, expression of proangiogenic growth factors like VEGF-B, VEGF-C, angiopoietin1, FGF1, FGF2, EGF, matrix metalloproteinase (MMP) 9, as well as PDGF-B has been reported in cardiac myocytes (Fig. 11.2). Expression of these molecules is triggered in cardiomyocytes by signaling molecules, transcription factors, and transcriptional co-regulators. In this regard, the transcription factor GATA4, which is activated by mechanical overload in cardiomyocytes, directly binds, and activates the Vegfa promoter [23]. Consequently, cardiomyocyte-specific GATA4 overexpression induces VEGF-A and capillary angiogenesis in the myocardium of mice, while in turn, genetic deletion of GATA4 in cardiac myocytes reduces myocardial angiogenesis and also leads to heart failure. In parallel, the hypoxia-sensitive transcription factor HIF1-α, which is a known direct regulator of VEGF-A and other angiogenic growth factors, becomes activated early in the course of cardiac pressure overload, when cardiomyocyte growth exceeds the ability of the existing capillary network to deliver enough oxygen for the muscle cells and hypoxia emerges [24]. Other cardiomyocyte-based regulators with positive effects on myocardial capillary growth include the transcriptional co-regulator peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC1α), the transcription factor signal transducer, and activator of transcription 3 (STAT3) and protein kinase B/Akt. Although capillary growth in the heart is enhanced during the initial compensatory phase of cardiac overload, capillary density decreases with disease progression, and capillary rarefaction is ultimately present in terminal heart failure. This might, at least in part, be due to the fact that cardiomyocyte GATA4 activation decreases and Hif1-α becomes inhibited by p53 in persisting pressure overload. Restitution of capillary density under these conditions, for example, by the delivery of angiogenic growth factors like VEGF-A and angiopoietin1, improves cardiac function, indicating that sufficient angiogenesis is crucial for the maintenance of heart function under pathological stress.
How do endothelial cells influence cardiomyocytes to maintain their function? First and foremost, they deliver oxygen and nutrients (i.e., amino acids, glucose, and fatty acids) to enable the production of ATP by myocytes. Second, paracrine factors released by the endothelial cells play an important role for cardiac homeostasis and survival. In a co-culture system of cardiomyocytes and endothelial cells, in which oxygen and nutrient delivery by capillaries naturally do not play a role, endothelial cells are essential for the survival of myocytes and also trigger their spatial organization and rhythmic contraction. As endothelial-derived paracrine factors, neuregulin1, which acts on ErbB2 and ErbB4 receptors on cardiomyocytes and which promotes myocyte survival and hypertrophy, as well as apelin1, which induces a strong positive inotropic response via its G-protein coupled receptor APJ, have been identified [2, 19]. Interestingly, both neuregulin1 and apelin1 are being evaluated as therapy in patients with heart failure.
11.3.3 Fibroblast-Cardiomyocyte Crosstalk
Cardiac fibroblasts are spindle-shaped cells that specifically express the PDGF receptor α as well as vimentin. They arise through EMT from the epicardium as well as the endocardium during embryonic development [25]. The primary function of cardiac fibroblasts is to synthesize (and degrade) extracellular matrix, which forms a three-dimensional structural network that supports cohesion of myocardial cells, cardiac shape, and function. Extracellular matrix (ECM) in the heart consists mainly of collagen I and III, fibronectin, proteoglycans, and glycoproteins [26]. In addition, fibroblasts communicate with cardiomyocytes via the release of specific growth factors, through extracellular matrix and even more directly by forming connexin containing gap-junction between these two different cell types.
In the embryonic heart, release of fibronectin, EGF-like growth factor and collagen by fibroblasts, promotes cardiac myocyte proliferation, by stimulating β1-integrin-dependent signaling in these cells [27]. In the healthy adult heart, fibroblasts are mainly quiescent, but become activated in response to mechanical overload and profibrotic molecules like TGFβ and connective tissue growth factor (CTGF), which are expressed in fibroblasts as well as cardiomyocytes (Fig. 11.2). Activation leads to a dramatic increase in fibroblast proliferation, secretion of extracellular matrix proteins, and growth factors. In addition, a fraction of fibroblasts (around 15 % in murine pressure overload) become myofibroblasts, which is a contractile cell type, characterized by the expression of α-smooth-muscle actin.
Co-culture with adult heart fibroblasts leads to hypertrophy in cardiomyocytes [27]. One of the growth factor responsible for this effect could be TGFβ1, which is released abundantly from myocytes as well as fibroblasts and which induces hypertrophy and dysfunction in cardiomyocytes and extracellular matrix production in fibroblasts. Interestingly, the endogenous prohypertrophic agonist angiotensin II primarily acts on the angiotensin type 1 receptor on cardiac fibroblasts and triggers cardiomyocyte growth indirectly through the induction of TGFβ1 and FGF2. FGF2 is mainly produced in myocardial fibroblasts and induces hypertrophy of adjacent cardiomyocytes. In support of this, FGF2 knockout mice showed reduced hypertrophy during pressure overload. Cardiac IGF1 is predominantly derived from fibroblasts, where its expression is induced by the transcription factor Krüppel-like-factor 5 (KLF5) [28]. Fibroblast IGF1 promotes cardiomyocyte hypertrophy and myocardial fibrosis, but is also essential for preventing heart failure and mortality during pressure overload in mice. Members of the IL-6 family like cardiotrophin-1 and leukemia inhibitory factor (LIF) are synthesized by cardiac fibroblasts and myocytes and signal through the transmembrane gp130 receptor to induce cardiomyocyte hypertrophy. In addition, CT-1 also promotes fibroblast migration, while LIF inhibits myofibroblast transition and collagen synthesis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


