Inotropic Agents
Ozlem Soran
Arthur M. Feldman
Oral and Intravenous Inotropic Agents
As noted elsewhere in this text, the primary hemodynamic abnormality in patients with congestive heart failure (CHF) secondary to the presence of a dilated cardiomyopathy is systolic dysfunction. Although dilated cardiomyopathic hearts also demonstrate some degree of diastolic dysfunction, it is the inability to maintain an appropriate cardiac output during stress or at rest that is responsible, in large part, for the symptoms of shortness of breath, edema, and fatigue that characterize patients with this disease. For this reason, physicians turned to inotropes-pharmacologic agents that augment ventricular function—as important therapeutic agents in the management of patients with both acute and chronic congestive heart failure.
Although these agents would intuitively be expected to improve cardiac output and, in so doing, ameliorate both the signs and symptoms of congestive heart failure, their use has been associated with long-standing controversy. Initially, investigators proposed that the use of an inotropic agent would be analogous to flogging a dead horse, as it was generally believed that the contractile apparatus could not be further stimulated because of irreversible structural and biological damage. More recently, the use of inotropic agents has been associated with increased mortality in a group of large, multicenter, randomized, and placebo-controlled clinical trials. However, it remains unclear as to whether an inotropic agent might have advantageous effects in specific subsets of patients or whether the deleterious effects of the inotropic agents developed to date are primarily caused by their ancillary properties rather than by their inherent ability to improve myocardial contractility.
Classification of Inotropic Agents
One dilemma in evaluating inotropic agents and their role in the therapy of CHF has been that all inotropic agents have been viewed in a generic sense. That is, regardless of mechanism of action or ancillary properties, all inotropic agents are thought to be equal. However, over the past two decades it has been clearly shown that myocardial contractility could be enhanced by activating or inhibiting a variety of biochemical pathways within the myocyte. In addition, our understanding of excitation—contraction coupling in both the normal and the failing heart has become far more sophisticated, leading to the identification of new and novel therapeutic targets. Finally, the use of recombinant molecular technology has led to the production of selective human proteins that can modify myocardial contractility. Thus, it now becomes important to classify inotropic agents according to their mechanisms of action and view them selectively rather than generically.
Initially, investigators classified inotropic agents according to their route of delivery (i.e., oral versus intravenous). As will be seen later in this chapter, although this classification was useful in describing existing agents it has not proven useful in classifying the many new and unique agents that have recently been developed. More recently, we proposed a method of classification based on the mechanism of action of the various agents (Table 29-1) (1). This allows the caregiver to correlate basic physiology with clinical actions, helps to clarify the potential synergistic
effects of some of the inotropic agents, and points out the class effects of some of these agents.
effects of some of the inotropic agents, and points out the class effects of some of these agents.
Table 29-1 Classes of Inotropic Agents by Mechanism of Action | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Inotropic Agents that Modulate Ion Channel Activity: the Cardiac Glycosides
Over 200 years ago, William Withering first noted that an extract of the foxglove plant was of benefit in the management of patients with CHF. In his initial report, Withering noted that the foxglove plant decreased edema in over 209 patients with dropsy (congestive heart failure) (2). However, subsequent reports by Bouillaud in 1835 (3) and Lewis in 1919 (4) suggested that digoxin was of benefit only in those patients with atrial fibrillation because of its ability to control heart rate in these patients. In contrast, Mackenzie and Christian (5,6) were early proponents of the use of digoxin in patients with normal sinus rhythm, publishing a review of the available literature in 1920. This controversy persisted into the 1990s and was fueled to some degree by reports noting an increase in mortality in patients receiving digoxin after suffering a myocardial infarction (MI) (7,8), as well as highly disparate reports regarding the clinical efficacy of digoxin (5,9,10,11,12,13). However, as noted later, recent large, multicenter, randomized, and placebo-controlled clinical trials have supported the benefits of digoxin in patients with CHF first noted by Withering two centuries ago.
The first conclusive demonstrations of the inotropic properties of digoxin came in 1927 when Wiggers and Stinson showed that digoxin could evoke a positive inotropic effect in the intact heart when heart rate and afterload were kept constant (14). Similarly, digoxin was shown to increase force generation in isolated cardiac muscle with expected shifts in the force-velocity relationships (15,16). More recently, studies in humans confirmed these initial findings in the laboratory, as digoxin enhanced myocardial contractility and increased stroke volume for a given preload and afterload (5). Of equal significance was that digoxin was also found to have a negative chronotropic effect as a result of parasympathetic activation and enhanced vagal tone, and also that it has anti-adrenergic properties (17,18). The effects of digoxin on cardiac contractility are generally accepted as resulting from increased availability of intracellular calcium (19). This increase in intracellular calcium concentration is secondary to inhibition of the sodium-potassium ATPase with a concomitant increase in cellular calcium transport in order to normalize electrolyte gradients (20). However, cardiac glycosides also have potent autonomic effects that can attenuate impaired arterial and cardiopulmonary baroreflex mechanisms in models of CHF (17).
The controversy surrounding the use of digoxin in the management of patients with CHF resulted partly from an inability to assay serum digoxin levels until 1969 (21). The ability to assay digoxin levels was important in understanding the clinical pharmacology of these products. Oral digoxin is absorbed by the gastrointestinal tract and excreted largely by the kidneys with a serum half-life of approximately 36 hours. Its excretion does not appear to be affected by either renal tubular absorption or secretion. However, approximately 20% to 25% of digoxin is protein-bound and, although digoxin is concentrated in the heart and kidneys, digoxin receptors are also present in skeletal muscle. Thus, patients with cardiac cachexia, the elderly, and those with renal dysfunction are prone to digoxin toxicity as a result of marked increases in digoxin concentrations (22). In addition, a variety of drugs and factors affecting gastrointestinal absorption can also alter the bioavailability of digoxin, including (a) cathartics and antacids (23); (b) severe diarrhea (24); (c) cholesterol-binding agents (25); (d) edema of the bowel wall, as is often seen in patients with right-sided heart failure (26,27); and (e) the presence of the intestinal bacterium Eubacterium lentum. Also, a variety of drugs and conditions can alter digoxin levels, including omeprazole (28); antibiotic therapy (29); electrolyte abnormalities including hyperkalemia or hypokalemia and hypernatremia or hyponatremia, indomethacin (30); cyclosporine, spironolactone (31); quinidine (32); verapamil (33); amiodarone (34); and propafenone (35). It is therefore imperative that digoxin doses be adjusted to account for the various pharmacologic and physiological factors that can influence digoxin levels, and that digoxin levels be monitored frequently during therapy.
Because of its relatively long half-life, many textbooks provide algorithms for loading digoxin either intravenously or orally to rapidly raise plasma digoxin levels without toxic effects. However, with the exception of patients in atrial fibrillation with rapid ventricular response, it is generally agreed that loading doses of digoxin are not necessary in the patient with CHF (36). Therefore, many physicians begin treatment empirically with 0.125 mg of digoxin per day with an increase to 0.250 mg per day if initial blood levels are low. Several studies have suggested a dose-response relationship between serum digoxin concentrations and hemodynamic effects (37,38). However, there is general agreement that the beneficial effects of digoxin can be obtained with a relatively low risk of toxicity when the serum level of digoxin is
between 1.0 and 1.5 ng/mL (36). Because serum digoxin levels equilibrate approximately 12 hours after an oral dose, many physicians recommend that their patients take their digoxin at night so that morning blood samples will accurately reflect peak drug levels.
between 1.0 and 1.5 ng/mL (36). Because serum digoxin levels equilibrate approximately 12 hours after an oral dose, many physicians recommend that their patients take their digoxin at night so that morning blood samples will accurately reflect peak drug levels.
The use of digoxin in patients with CHF should always be undertaken with a clear recognition of the potential toxicities associated with its use. Clinically, digoxin toxicity can be divided into three major categories: gastrointestinal, neurologic, and cardiovascular. Gastrointestinal effects may include nausea, anorexia, abdominal pain, diarrhea, and vomiting (39). Neurologic toxicities include fatigue, headache, fatigue, visual changes, confusion, psychosis, and seizures; however, visual changes including halos around lights or enhanced yellow and green hues are almost pathognomonic for digoxin toxicity (40). Cardiovascular toxicities of digoxin are also diverse and may include virtually any type of arrhythmia or conduction disturbance (41). However, suspicion of digoxin toxicity should be heightened by the appearance of regularization of an irregular rhythm or a new bradyarrhythmia or tachyarrhythmia. The recent introduction of digoxin-specific antibody fragments has had a major impact on the therapy of digoxin toxicity (42). In a study of 150 patients with life-threatening digoxin intoxication, the use of digoxin-specific Fab antibody fragments was effective in over 90% of the patients, with adverse effects in fewer than 1% of patients.
A decade ago, a group of clinical studies evaluated the efficacy of digoxin in randomized studies. In a double-blind, placebo-controlled comparison of digoxin and the adrenergic agonist xamoterol (433 patients with chronic heart failure), digoxin was not effective in improving exercise duration, although it was associated with improvement in the tiredness associated with daily life as measured by the Likert scale (43). However, these patients were not well-characterized, as nearly 25% of the patients were in New York Heart Association (NYHA) Class I CHF, and the etiology of heart failure was not defined in a large number of the patients. In the Captopril-Digoxin Multicenter Research Group study, patients receiving diuretics were randomized to receive either captopril or digoxin (44). Digoxin treatment increased ejection fraction compared with captopril and placebo and decreased hospitalizations when compared with placebo. However, only captopril improved NYHA class or exercise time and digoxin was associated with an increase in ventricular ectopy.
Finally, DiBianco et al. compared oral milrinone, digoxin, and their combination in the treatment of patients with chronic heart failure (45). Treatment with digoxin significantly increased treadmill exercise time, reduced the frequency of decompensation from heart failure, and increased ejection fraction. However, it should be noted that a substantial number of patients failed to meet the entry criteria because of an inability to exercise. Although these studies suggested a beneficial effect of digoxin, they were not consistent in terms of either outcomes or patient enrollment. Furthermore, the studies were limited by an absence of therapy with angiotensin-converting enzyme (ACE) inhibitors, agents subsequently shown to improve survival and limit hospitalizations in this patient population (46,47).
Over the past decade, several seminal and large clinical trials have clearly demonstrated the utility of digoxin in the therapy of patients with CHF who were receiving concomitant therapy with an ACE inhibitor. In the RADIANCE study (Randomized Assessment of Digoxin on Inhibitors of the ANgiotensin-Converting Enzyme), patients with Class II or III heart failure and left ventricular ejection fractions of 35% or less who were receiving therapy with an ACE inhibitor and digoxin were randomized to having their digoxin continued or being switched to placebo (48). In these patients who were clinically stable while receiving diuretics, digoxin, and an ACE inhibitor, the withdrawal of digoxin was associated with worsening heart failure, deterioration in functional capacity, worsening quality of life, decreased ejection fractions, and increases in heart rate and body weight. In the most definitive assessment of the role of digoxin in the management of patients with chronic CHF, 6,800 patients with left ventricular ejection fractions of <45% and signs and symptoms of CHF were randomized to receive either digoxin or placebo (49). Physicians were strongly encouraged to treat patients with ACE inhibitors, and 94% of patients were receiving these medications; 81% of patients were receiving diuretics. When the primary endpoint of mortality was assessed, randomization to active therapy had no effect on survival over 52 months of follow-up. However, the use of digoxin was associated with 6% fewer hospitalizations, fewer hospitalizations for worsening heart failure, and a trend toward a decrease in the risk of death attributable to worsening heart failure (p = 0.06). In an ancillary trial, digoxin had similar effects in patients with dilated hearts but with ejection fractions >45%. Thus, the results of this study suggest that in clinical practice digoxin therapy is likely to affect the frequency of hospitalizations without adversely affecting survival.
Several recent studies have strongly influenced our views regarding the role of digoxin in the treatment of heart failure. First, it has been recognized that serum digoxin concentrations in the therapeutic range (up to 2 ng per mL) could be associated with an increased frequency of hospitalizations for cardiovascular events other than heart failure and an increased risk of death due to arrhythmias or MI (50). Thus, physicians now recognize that digoxin doses that had previously been considered to be safe may adversely affect the heart, and some clinicians recommend that the digoxin dose be less than 1 ng per mL (51,52). Furthermore, heightened concern exists that digoxin may be contraindicated in patients who are post-MI and those with ongoing ischemia (53). Finally, a retrospective analysis of the Digoxin Investigation Group (DIG) trial suggested that while digoxin had modest effects in men, it had potentially adverse effects in women (54). Thus, because physicians now have an expanded armamentarium for the treatment of heart failure that includes agents such as β-blockers, ACE inhibitors, and aldosterone antagonists as well as new devices, enthusiasm for the use of digoxin has waned and its use has diminished. However, in patients who remain symptomatic despite optimal pharmacologic therapy, digoxin may still play a role in heart failure management.
It is important to note that digoxin has no beneficial properties in patients with diastolic dysfunction and its
role in patients with minimal systolic function remains undefined. In addition, it is unclear whether patients who are asymptomatic but have dilated cardiomyopathies are candidates for receiving digoxin therapy. It should also be recognized that because of its positive inotropic properties, digoxin is contraindicated in patients with symptoms of heart failure secondary to hypertrophic cardiomyopathy, as it can worsen pre-existing outflow tract obstructions and increase left ventricular end-diastolic pressures.
role in patients with minimal systolic function remains undefined. In addition, it is unclear whether patients who are asymptomatic but have dilated cardiomyopathies are candidates for receiving digoxin therapy. It should also be recognized that because of its positive inotropic properties, digoxin is contraindicated in patients with symptoms of heart failure secondary to hypertrophic cardiomyopathy, as it can worsen pre-existing outflow tract obstructions and increase left ventricular end-diastolic pressures.
Adrenergic Agonists and the Management of Patients with Congestive Heart Failure
The human myocardium relies on β-adrenergic stimulation to augment contractility when the need arises for increased cardiac output such as during exercise or stress (Fig. 29-1) (55,56). This is in contrast to many laboratory animal species such as the rat and the guinea pig, in which the heart is predominantly β/α1– or H2-histaminic, respectively (57,58,59). In addition, unlike many animal species, the human heart contains a balance of β1– and β2-adrenergic receptors that demonstrate marked differences in pharmacologic specificity and regulatory behavior (60,61). The human myocardium also contains a relatively small number of α1-adrenergic receptors, which mediate inconsistent inotropic effects in isolated tissues (10). However, the α1– receptors are coupled to myocardial cell growth in model systems and this receptor pathway may play a role in the development of cellular hypertrophy that accompanies the development of cardiac dilation and failure. Recent studies have identified the presence of β3-adrenergic receptors; however, their role in the regulation of myocardial function has not yet been clearly defined (62,63,64).
The β-adrenergic receptor-coupled pathways have been extensively studied in both the normal and failing human
heart over the past two decades (65,66,67). It is now well-documented that, under normal circumstances, myocardial contractility is augmented through activation of the β-adrenergic—G-protein—adenyl cyclase complex (RGC). This complex serves to identify, transduce, and amplify extracellular signals by increasing the production of the intracellular second messenger cyclic adenosine mono-phosphate (cAMP). When a ligand interacts with a β-adrenergic receptor, the affinity of the α subunit of the G stimulatory protein (Gsα) for guanosine 5′-triphosphate (GTP) is enhanced, facilitating exchange of guanosine 5′-diphosphate (GDP) for GTP. When GDP is released from the guanine nucleotide-binding site of Gs, the G-protein heterotrimer dissociates, releasing free subunits and Gsα-GTP. This free Gsα is able to activate the catalytic subunit of adenyl cyclase, resulting in increased production of cAMP. cAMP in turn activates cAMP-dependent protein kinases, which serve as relatively promiscuous phosphorylators that can regulate a variety of cellular proteins including phospholamban, troponin, and the gated calcium channel, resulting in enhanced contraction and relaxation. Alternatively, cAMP can be metabolized to an inactive product by the enzyme phosphodiesterase (PDE). The RGC signal transduction system is under the inhibitory control of the inhibitory guanine nucleotide-binding protein Gi. Activation of Gi inhibits adenylyl cyclase activity not so much by a direct action on the catalytic subunit but, rather, by shifting the stoichiometry of G-protein binding such that there is less free Gsα-GTP to activate the catalytic subunit of adenyl cyclase (5).
heart over the past two decades (65,66,67). It is now well-documented that, under normal circumstances, myocardial contractility is augmented through activation of the β-adrenergic—G-protein—adenyl cyclase complex (RGC). This complex serves to identify, transduce, and amplify extracellular signals by increasing the production of the intracellular second messenger cyclic adenosine mono-phosphate (cAMP). When a ligand interacts with a β-adrenergic receptor, the affinity of the α subunit of the G stimulatory protein (Gsα) for guanosine 5′-triphosphate (GTP) is enhanced, facilitating exchange of guanosine 5′-diphosphate (GDP) for GTP. When GDP is released from the guanine nucleotide-binding site of Gs, the G-protein heterotrimer dissociates, releasing free subunits and Gsα-GTP. This free Gsα is able to activate the catalytic subunit of adenyl cyclase, resulting in increased production of cAMP. cAMP in turn activates cAMP-dependent protein kinases, which serve as relatively promiscuous phosphorylators that can regulate a variety of cellular proteins including phospholamban, troponin, and the gated calcium channel, resulting in enhanced contraction and relaxation. Alternatively, cAMP can be metabolized to an inactive product by the enzyme phosphodiesterase (PDE). The RGC signal transduction system is under the inhibitory control of the inhibitory guanine nucleotide-binding protein Gi. Activation of Gi inhibits adenylyl cyclase activity not so much by a direct action on the catalytic subunit but, rather, by shifting the stoichiometry of G-protein binding such that there is less free Gsα-GTP to activate the catalytic subunit of adenyl cyclase (5).
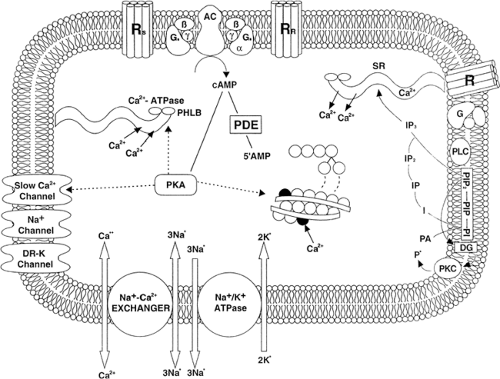 Figure 29-1 Biochemical pathways important in the regulation of cardiac contractility. AC, adenylyl cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; DG, diacylglycerol; DR, delayed rectifier; G, guanine nucleotide-binding regulatory proteins that may stimulate (αGs) or inhibit (αGi) adenylyl cyclase; I, inositol; IP, inositol phosphate; IP2, inositol bisphosphate; IP3, inositol trisphosphate; PDE, phosphodiesterase; PHLB, phospholamban; PI, phosphatidylinositol; PIP, phosphatidylinositol 4-phosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; R, sarcolemmal receptor; Ri, inhibitory receptor; Rs, stimulatory receptor; SR, sarcoplasmic reticulum; Tn, troponin. |
The RGC complex further modifies cellular function through direct effects of the G-protein subunits on selected ion channels (60). Furthermore, recent studies have demonstrated the importance of other regulatory proteins in the normal functioning of the RGC complex. For example, a family of β-adrenergic receptor kinases (β-ARK) are expressed in the heart and preferentially phosphorylate the agonist-occupied form of the β-adrenergic receptor independent of cAMP (68,69,70). In so doing, these G-protein-coupled receptors are able to modify the effectiveness of adrenergic agonists by desensitizing the β1-adrenergic receptors (70). The role of the β-ARKs in cardiac physiology has been validated in transgenic mice overexpressing β-ARK1 (71). These mice express threefold to fivefold more β-ARK1 activity than wild type mice and demonstrate significant attenuation of isoproterenol-induced inotropy.
Further support for the role of the RGC complex in maintaining cardiac function comes from studies using transgenic mice. Overexpression of the β2-adrenergic receptor results in increased basal myocardial adenyl cyclase activity, enhanced atrial contractility, and increased left ventricular function in vivo, changes that were virtually identical to those seen when wild type mice were exposed to maximal concentrations of adrenergic agonists (72). These mice also demonstrate markedly enhanced relaxation that is associated with a selective reduction in the amount of phospholamban protein (73). Consistent with these findings, transgenic mice overexpressing β-ARK demonstrated attenuation of isoproterenol-stimulated left ventricular contractility, dampening of myocardial adenylyl cyclase activity, and reduced functional coupling of the β-adrenergic receptors, whereas mice expressing the β-ARK inhibitor displayed enhanced cardiac contractility in the presence or absence of isoproterenol (71). In contrast, Overexpression of the β1-adrenergic receptor has been reported to effect the development of a dilated cardiomyopathy. However, the heart failure phenotype does not occur until the mice are well over 1 year of age (M. R. Bristow et al., personal communication). Similarly, overexpression of the α subunit of Gs results in the development of cardiac dilation and diminished left ventricular function (74). However, as noted with β1-adrenergic receptor overexpression, these changes are seen only in older mice (75).
It is important to note that a characteristic feature of heart failure in humans is the presence of marked and substantial changes in the expression and function of many (if not most) of the key components of the RGC complex. In 1982, Ginsburg et al. first noted that papillary muscle isolated from failing human heart was less responsive to isoproterenol than was heart obtained from nonfailing controls (76). This diminished sensitivity occurred in the presence of increased concentrations of plasma and cardiac norepinephrine (77) and was presumably a result of downregulation of the adrenergic receptors (78). This hypothesis was supported by the seminal study of Bristow et al., who found that heart failure in humans was associated with profound downregulation of the β-adrenergic receptor (79). This downregulation was specific and selective for the β1-adrenergic receptor, resulting in a shift in the β1/β2 receptor ratio (80) and was caused by a diminution in the expression of β1-adrenergic receptor mRNA rather than by sequestration within the sarcolemma (81). Similar changes in receptor-effector coupling occur in the aged myocardium (82,83). Interestingly, in some patients with idiopathic dilated cardiomyopathy, the extracellular loop of the β1-adrenergic receptor serves as an autoimmune epitope, which might have physiological implications (84). In addition, heart failure in humans is associated with abnormalities in the functional activity of the G-inhibitory-protein, resulting in diminished receptor-effector coupling (85,86,87,88). More recently, investigators have noted altered expression of β-ARK in the failing human heart, suggesting that altered phosphorylation may also play a role in receptor desensitization (89,90).
In view of the marked abnormalities in RGC coupling that characterize the failing human myocardium, it had been proposed that the use of positive inotropic agents having effects through various components of the RGC pathway would have only deleterious consequences. Not only were these pathways less sensitive to adrenergic stimulation, but continued production of cAMP could further compromise function or be cardiotoxic. Although this hypothesis was intuitively reasonable, several findings raised questions as to the potential efficacy of RGC-directed inotropic therapy. First, administration of adrenergic agonists has been associated with abrupt withdrawal of adrenergic drive in patients with symptomatic CHF (91). In addition, when β-receptor density was compared between patients who had received high doses of dobutamine for long periods of time before transplantation and those who had received short periods of dobutamine infusion at relatively low concentrations before transplantation, the higher dose and duration of β-adrenergic therapy were actually associated with higher numbers of β-adrenergic receptors
(59). Finally, when intracellular concentrations of cAMP are increased by inhibiting the function of PDE, there is no associated decrease in β-adrenergic receptor density despite a marked improvement in myocardial contractility (92). When adrenergic drive is relatively low, PDE inhibitors might increase contractility to a limited degree. However, when the RGC complex is activated during exercise or by administration of exogenous catecholamines, the presence of a PDE inhibitor might act synergistically to improve myocardial contractility, as seen in the discussion later.
(59). Finally, when intracellular concentrations of cAMP are increased by inhibiting the function of PDE, there is no associated decrease in β-adrenergic receptor density despite a marked improvement in myocardial contractility (92). When adrenergic drive is relatively low, PDE inhibitors might increase contractility to a limited degree. However, when the RGC complex is activated during exercise or by administration of exogenous catecholamines, the presence of a PDE inhibitor might act synergistically to improve myocardial contractility, as seen in the discussion later.
Clinical Use of Adrenergic Agonists
Dobutamine
Clinicians first used the adrenergic agonist isoproterenol to treat patients with cardiogenic shock in the early 1960s. However, its use was limited because of associated tachycardia, arrhythmias, and diminished vascular resistance (93). In 1975, Tuttle and Mills developed the adrenergic agonist dobutamine by modifying the amino-terminal end of isoproterenol (94). This modification retained the potent inotropic properties of isoproterenol without the associated tachycardia and arrhythmias, although it was useful only as an intravenous preparation. Dobutamine has β1, β2, and α1 effects (95); however, its ability to affect contractility without having positive chronotropic effects may lie in the unique isomeric structure of dobutamine (95,96). Because both β2-and α1-receptors are found in the peripheral vasculature, dobutamine elicits a relatively balanced effect on the peripheral vasculature. However, its beneficial effects in patients with CHF might derive from a complex relationship among the various receptors with which it interacts.
There is a general consensus that dobutamine can effect a rapid and relatively robust increase in cardiac contractility in the normal heart as well as in the failing human heart, although the response in patients with CHF is clearly blunted (Table 29-2) (97). Indeed, peak effects can be obtained in as little as 10 minutes and there is a direct correlation between dobutamine dose, plasma concentration, and hemodynamic response (98). The short half-life of dobutamine facilitates its use in clinically unstable patients, as undesirable side effects (including tachyarrhythmias, headaches, anxiety, excessive changes in blood pressure, or increased rates of ventricular response in patients with atrial fibrillation) can be reversed in as little as 10 minutes after discontinuation of intravenous therapy.
In clinical use, dobutamine has been shown to reduce ventricular filling pressures, increase stroke volume, and enhance cardiac output (99,100,101,102,103,104). Although the continuous use of dobutamine might be associated with receptor down-regulation, the efficacy of prolonged therapy remains somewhat controversial. Unverferth et al. demonstrated the development of tolerance in patients receiving dobutamine for over 96 hours (105). However, tolerance could be abrogated by simply increasing infusion rates. More importantly, patients demonstrated an increase in both cardiac output and coronary blood flow that was accompanied by an increase in serum sodium, a decrease in blood urea nitrogen, and an increase in the ATP/creatine phosphate ratio. Furthermore, as mentioned earlier, studies by both Colucci and Bristow noted that the improvements in myocardial function that were effected by continuous infusion of dobutamine actually decreased adrenergic drive and its consequences on adrenergic receptor density.
Although the usefulness of dobutamine in the treatment of patients with acute exacerbation of CHF is well-recognized, several recent reports have questioned whether dobutamine and intravenous diuretics should be the first agents used in these patients, or whether vasodilators in combination with intravenous diuretics are more efficacious (106,107,108). Furthermore, it is unclear whether patients receiving inotropic and/or intravenous vasodilator therapy should undergo placement of a pulmonary artery catheter to facilitate titration of the medications (109). This important question will be addressed by the upcoming ESCAPE trial under the sponsorship of the National Heart, Lung, and Blood Institute (NHLBI).
Although there has been a general consensus regarding the use of dobutamine in patients with acute heart failure, the usefulness of either intermittent or chronic dobutamine in patients with end-stage heart failure has been a highly contentious issue. Intermittent therapy with dobutamine was based in large part on the finding that the beneficial effects of a 72-hour dobutamine infusion in patients with chronic CHF persisted for up to 4 weeks in over 50% of patients after discontinuation of dobutamine therapy (97). Dobutamine effected a sustained improvement in hemodynamic indexes as well as in biochemical and ultrastructural parameters of the heart, leading investigators to propose that intermittent therapy might provide long-term benefits for patients with
chronic heart failure (105,110). Indeed, a group of investigations in animal models of CHF (111,112) as well as several clinical studies (113,114) supported this hypothesis. Enthusiasm for the use of intermittent dobutamine was dampened, however, with the presentation of a report by Dies that demonstrated an increase in early mortality in patients treated with intermittent dobutamine (115). However, this study was highly flawed in that (a) electrolyte levels were not routinely measured; (b) high concentrations of dobutamine were used throughout the study; and (c) dobutamine concentrations were not titrated for interstudy changes in body weight. Despite this report, several centers have continued to report favorable effects of chronic dobutamine therapy in patients with chronic heart failure (116,117).
chronic heart failure (105,110). Indeed, a group of investigations in animal models of CHF (111,112) as well as several clinical studies (113,114) supported this hypothesis. Enthusiasm for the use of intermittent dobutamine was dampened, however, with the presentation of a report by Dies that demonstrated an increase in early mortality in patients treated with intermittent dobutamine (115). However, this study was highly flawed in that (a) electrolyte levels were not routinely measured; (b) high concentrations of dobutamine were used throughout the study; and (c) dobutamine concentrations were not titrated for interstudy changes in body weight. Despite this report, several centers have continued to report favorable effects of chronic dobutamine therapy in patients with chronic heart failure (116,117).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


