Inhibition of the Renin-Angiotensin-Aldosterone System in Chronic Heart Failure: Rationale, Results, and Current Recommendations
John R. Teerlink
Kiran K. Khush
Barry M. Massie
Our understanding of the pathophysiology of chronic heart failure has progressed significantly, from simple pump failure, to an expanded cardio-renal model, to our currently accepted neurohormonal model (1). We now recognize the vital role that neurohormonal activation plays in the initiation and progression of heart failure (HF), even in the absence of further direct insults. The renin-angiotensin-aldosterone system (RAAS) lies at the heart of this neurohormonal cascade, and our understanding of the complex, important roles played by angiotensin II and aldosterone continues to evolve. While the hemodynamic and renal effects of these hormones are now well-characterized, their cellular and molecular effects continue to be a source of intense investigation.
Clinical observations, leading to clinical trials using a variety of neurohormonal antagonists which have improved symptoms and outcomes of HF patients, have given direction to basic investigations clarifying the physiological and cellular mechanisms by which these endogenous neurohormones and their antagonists produce their deleterious and beneficial effects. This chapter will review the role of the RAAS in the pathophysiology of HF, with an emphasis on the rationale for the use of RAAS inhibitors, clinical trial, results, and current treatment recommendations.
Rationale for Blockade of the Renin-Angiotensin-Aldosterone System in Heart Failure
Hemodynamic Alterations in Heart Failure
The characteristic hemodynamic abnormalities in patients with HF are a reduction in stroke volume and cardiac output, and concomitant elevation of left and right ventricular filling pressures, either at rest or with activity (2). These changes are accompanied by an increase in systemic vascular resistance and redistribution of regional blood flow, thereby contributing to the typical signs and symptoms of HF, such as fluid retention and inadequate blood flow to vital organs including the kidneys and exercising muscle. Such physiological alterations result in dyspnea, exercise intolerance, fatigue, and edema. Neurohormonal activation plays a central role in causing these abnormalities,
which in turn can be alleviated by blocking these responses.
which in turn can be alleviated by blocking these responses.
Neurohormonal Activation in Heart Failure
HF represents a highly complex syndrome in which myocardial injury leads to neurohormonal activation and subsequent ventricular remodeling (3) (Fig. 26-1). In the neurohormonal model, HF develops as a result of the overexpression of peptide molecules which exert toxic long-term effects on the cardiovascular system (1,4,5). In the acute phase of HF, neurohormonal activation may help maintain adequate cardiac output and peripheral perfusion. Sustained neurohormonal activation, however, eventually results in increased cardiac wall stress, ventricular dilatation, and adverse remodeling (6,7). A variety of endogenously produced substances, including norepinephrine, angiotensin II, aldosterone, endothelin, and tumor necrosis factor-alpha (TNF-α), have been implicated as biologically active molecules which contribute to disease progression in the failing heart.
Therefore, although the early approaches to treating HF, such as positive inotropic agents and diuretics, provide symptomatic relief and hemodynamic improvement, they have relatively limited impact on its natural history. In contrast, inhibition of the activated RAAS and sympathethetic nervous system prevents or slows ventricular remodeling- a process closely linked to the progression of HF, discussed elsewhere in this text-and consequently has beneficial effects on its natural history
The Endocrine Renin-Angiotensin-Aldosterone System
As described in another chapter of this text, the activation of the RAAS in acute and chronic HF is triggered by reduced stroke volume or cardiac output, with resultant impairment of renal blood flow and perfusion pressure. The cascade is initiated by the release of renin from the juxtaglomerular cells of the kidney in response to renal hypoperfusion and sympathetic activation (8). Angiotensinogen produced by the liver is then cleaved by renin to yield the inactive decapeptide angiotensin I (Ang I). Circulating Ang I is then converted to angiotensin II (Ang II) in the lungs and other tissues by angiotensin-converting enzyme (ACE). Ang II exerts its biological effects by binding to Ang II receptors in target organs and tissues (Fig. 26-2).
In the kidneys, Ang II causes sodium and water retention and vasoconstriction of efferent arterioles of the glomerulus, thereby increasing blood pressure and sodium reabsorption. Ang II concomitantly stimulates the secretion of aldosterone by the adrenal cortex and arginine vasopressin by the posterior pituitary gland, both of which contribute to extracellular volume expansion and sympathetic activation. At a molecular level, Ang II stimulates myocyte hypertrophy, apoptosis, fibroblast proliferation, collagen deposition, and interstitial fibrosis (7). Furthermore, evidence now implicates the RAAS in the development of atherosclerosis (9), which indirectly may negatively impact on the natural history of HF patients. These disparate effects, in sum, promote the development of left ventricular dysfunction, vascular abnormalities, and the clinical syndrome of HF (Fig. 26-3).
The Cardiac Renin-Angiotensin-Aldosterone System
Until relatively recently, the classical endocrine RAAS was considered to be the mediator of the cardiovascular effects of angiotensin and aldosterone. However, experimental evidence now provides strong support for the existence of a local cardiac RAAS (10,11). The individual components of the RAAS are present within cardiac and vascular myocytes and fibroblasts, leading to production of neurohormones such as Ang II and aldosterone within the myocardium itself. These mediators act in an autocrine and paracrine manner, independent of the effects of circulating renin, thereby contributing to local cardiac injury.
The discovery of local expression and activation of the RAAS in both the myocardium and vasculature has prompted investigation into the tissue-specific affinity of different ACE inhibitors (ACEIs), and whether tissue-specificity confers any clinical benefit. Although some ACEIs have a higher tissue affinity than others (12,13), there is no evidence that the commonly used agents differ in their clinical effects and the relative clinical importance of the cardiac tissue RAAS remains unclear.
Angiotensin-Converting Enzyme
ACE is a zinc metallopeptidase that catalyzes the conversion of Ang I to Ang II. This enzyme, which is primarily tissue-based, is induced in virtually all states of cardiac injury, including volume overload (14,15), myocardial
infarction (MI) (16,17), and HF. Its actions, which are triggered by elevated left ventricular wall stress, regulate the balance between competing vasoconstrictive and vasodilatory systems. While the RAAS causes vasoconstriction and salt retention via the actions of Ang II and aldosterone, the kallikrein-kinin system leads to vasodilation and natriuresis through the effects of bradykinin (18). Bradykinin, which is degraded by ACE (also known as kininase II), is a potent vasodilator that stimulates endothelial cells to release prostaglandins, which in turn enhance nitric oxide production (19). Reduced prostaglandin release may thereby adversely affect the clinical outcomes of HF patients. Inhibition of ACE ultimately slows the progression of cardiovascular diseases by improving endothelial function, exerting antiproliferatory and antimigratory effects on smooth muscle cells, neutrophils, and monocytes (20,21), and by exerting antithrombotic effects (13).
infarction (MI) (16,17), and HF. Its actions, which are triggered by elevated left ventricular wall stress, regulate the balance between competing vasoconstrictive and vasodilatory systems. While the RAAS causes vasoconstriction and salt retention via the actions of Ang II and aldosterone, the kallikrein-kinin system leads to vasodilation and natriuresis through the effects of bradykinin (18). Bradykinin, which is degraded by ACE (also known as kininase II), is a potent vasodilator that stimulates endothelial cells to release prostaglandins, which in turn enhance nitric oxide production (19). Reduced prostaglandin release may thereby adversely affect the clinical outcomes of HF patients. Inhibition of ACE ultimately slows the progression of cardiovascular diseases by improving endothelial function, exerting antiproliferatory and antimigratory effects on smooth muscle cells, neutrophils, and monocytes (20,21), and by exerting antithrombotic effects (13).
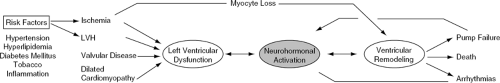 Figure 26-1 Pathophysiology of heart failure. Neurohormonal activation, through the reninangiotensin-aldosterone system and other sympathetic axes, is central to the development and progression of heart failure. LVH, left ventricular hypertrophy. |
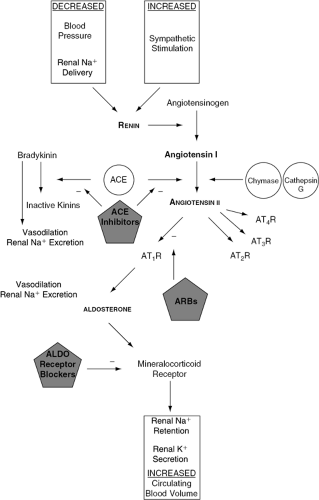 Figure 26-2 The renin-angiotensin-aldosterone system, including sites of pharmacologic inhibition of this neurohormonal axis. Na+, sodium; K+, potassium; ACE, angiotensin-converting enzyme; ATR, angiotensin receptor; ARB, angiotensin receptor blocker, ALDO, aldosterone; BK, bradykinin. |
Pharmacology of Renin-Angiotensin-Aldosterone System Manipulation
The introduction of the first oral ACEIs in the 1980s represented a major breakthrough in the management of HF
patients. Several large, randomized, placebo-controlled studies in patients with left ventricular systolic dysfunction, treated with diuretics, proved that this class of drugs can improve symptoms and decrease morbidity and mortality in patients with mild, moderate, or even severe HF, regardless of its etiology. These drugs, which block the conversion of Ang I to Ang II, decrease the formation of Ang II and the Ang II-mediated release of aldosterone. ACE inhibitors also inhibit the breakdown of bradykinin, resulting in increased plasma concentrations of nitric oxide and vasodilating prostaglandins (22). Inhibition of Ang II and aldosterone production eventually leads to a decrease in sodium and water retention and adverse cardiac remodeling. Cardiac preload and afterload are concomitantly reduced, leading to an improvement in hemodynamic status (23).
patients. Several large, randomized, placebo-controlled studies in patients with left ventricular systolic dysfunction, treated with diuretics, proved that this class of drugs can improve symptoms and decrease morbidity and mortality in patients with mild, moderate, or even severe HF, regardless of its etiology. These drugs, which block the conversion of Ang I to Ang II, decrease the formation of Ang II and the Ang II-mediated release of aldosterone. ACE inhibitors also inhibit the breakdown of bradykinin, resulting in increased plasma concentrations of nitric oxide and vasodilating prostaglandins (22). Inhibition of Ang II and aldosterone production eventually leads to a decrease in sodium and water retention and adverse cardiac remodeling. Cardiac preload and afterload are concomitantly reduced, leading to an improvement in hemodynamic status (23).
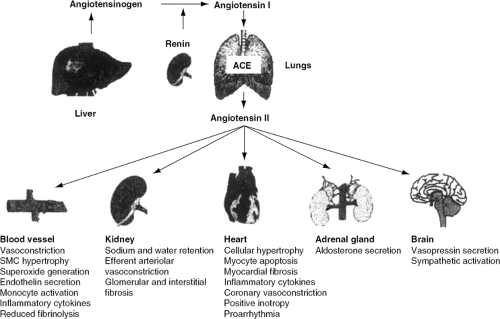 Figure 26-3 Pathophysiology of the renin-angiotensin-aldosterone system, demonstrating the end-organ effects of Ang II. (Reproduced from Givertz MM. Manipulation of the renin-angiotensin system. Circulation. 2001;104:e14-e18 , with permission.) SMC, smooth muscle cell. |
Angiotensin-Converting Enzyme Inhibitors
Angiotensin-Converting Enzyme Inhibitors: Hemodynamic Effects
The initial studies with ACEIs focused on their hemodynamic effects (24,25,26,27). These studies demonstrated significant decreases in mean arterial pressure and systemic vascular resistance, increases in cardiac output and stroke volume, and reductions in left and right ventricular filling pressures, as well as end-diastolic volumes. Similar changes were noted during exercise. Interestingly, rather than promoting a reflex tachycardia, ACEIs were shown to cause a modest decrease in heart rate (24). This effect may be due to a withdrawal of sympathetic tone in the setting of ACEI therapy. Importantly, the hemodynamic effects of ACEIs were maintained during chronic therapy. Sustained hemodynamic effects, as well as increased exercise capacity, maximal work load, and oxygen consumption were observed following 3 months of treatment with captopril, compared to those who were given placebo (Table 26-1) (27).
Angiotensin-Converting Enzyme Inhibitors and Vascular Protection
The importance of ACE inhibition on prevention of vascular events was initially demonstrated in several large, placebo-controlled trials which enrolled patients with HF and myocardial infarction (MI) (28,29). Patients receiving ACEIs had an unexpected 11% to 25% lower rate of recurrent MI compared to placebo. This led to the vascular hypothesis, which suggests a preventive effect of ACEIs in a wide variety of patients with a history of stroke or transient ischemic attacks, coronary artery disease, peripheral arterial disease, diabetes mellitus, renal disease, and hypertension with additional risk factors, even in the absence of left ventricular dysfunction (Fig. 26-4).
Table 26-1 Effects of Ace Inhibitors on Hemodynamics and Left Ventricular Remodeling | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
Angiotensin-Converting Enzyme Inhibitors in Chronic Heart Failure
In the first multicenter, randomized, placebo-controlled clinical trial of an ACEI, the Captopril Multicenter study (26), 92 patients with HF refractory to digitalis and diuretics were randomized to captopril or placebo. After 12 weeks, 80% of the patients treated with captopril experienced a clinical improvement, compared to 27% of those given placebo. The captopril group demonstrated a significant improvement in New York Heart Association (NYHA) functional class, a 24% increase in exercise tolerance, and a significant increase in left ventricular ejection fraction (LVEF) when compared to placebo. The improvement seen in exercise tolerance was gradual and sustained throughout the follow-up period (26). Albeit small, this study initiated the era of ACEI therapy for chronic HF.
The next major breakthrough in HF therapy was the demonstration that ACEIs not only improve hemodynamics and symptoms but also long-term prognosis. In a series of trials, ACEIs were shown to decrease morbidity and mortality in patients with mild, moderate, and severe symptoms, regardless of HF etiology (26,30,31,32). The first mortality trial was CONSENSUS I (Cooperative North Scandinavian Enalapril Survival Study), in which patients with severe NYHA Class IV HF were randomized to enalapril or placebo. After 20 months, the patients treated with enalapril experienced a 27% reduction in overall mortality and a 50% decrease in mortality due to progressive HF (33) (Fig. 26-5). The SOLVD (Studies of Left Ventricular Dysfunction) Treatment trial extended the indications for ACEI therapy by demonstrating a significant mortality benefit in patients with less-severe HF (NYHA Class II-III) than those enrolled in CONSENSUS I (Fig. 26-6) (34).
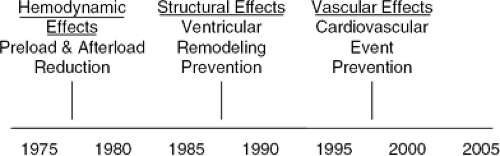 Figure 26-4 Evolving paradigms for the use of angiotensin-converting enzyme inhibitors for the treatment of heart failure. |
A systematic review of the trials of ACEI therapy in patients with chronic HF pooled the data from 7,105 patients enrolled in 32 randomized, controlled studies which remain the primary source of data on these agents (35). This comprehensive analysis demonstrated a 23% reduction in mortality and a 35% reduction in the combined endpoint of mortality or hospitalization for HF in patients treated with ACEIs (Table 26-2). The reduction in mortality was primarily due to fewer deaths from progressive HF, and patients with poor LV function appeared to
gain the most benefit. This review also suggested that, while the greatest effects were seen during the initial 3 months of ACEI therapy, additional benefits accrued during prolonged treatment.
gain the most benefit. This review also suggested that, while the greatest effects were seen during the initial 3 months of ACEI therapy, additional benefits accrued during prolonged treatment.
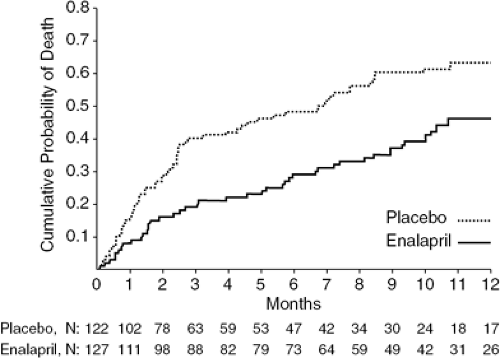 Figure 26-5 Kaplan-Meier survival curve from the CONSENSUS study. (Reproduced from The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study [CONSENSUS]. N Engl J Med. 1987;316:1429–1435 , with permission.) |
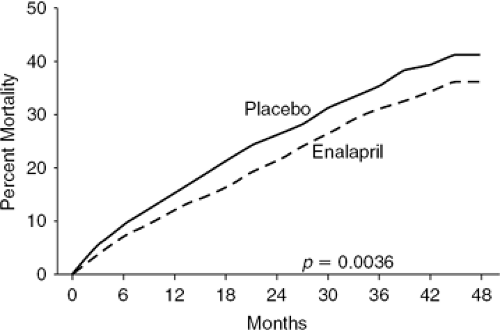 Figure 26-6 Kaplan-Meier survival curve from SOLVD-Treatment study. (Reproduced from the SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302 , with permission.) |
Importantly, ACEI therapy proved superior to treatment with other vasodilators, although their hemodynamic effects may be similar. In the V-HeFT II trial, treatment with enalapril reduced mortality by 28% compared to combination therapy with isosorbide dinitrate and hydralazine (36). These findings suggest that neurohormonal antagonism with RAAS blockers favorably influences the natural history of HF and has significant beneficial effects beyond hemodynamic improvement alone.
Angiotensin-Converting Enzyme Inhibitors: Effects on Left Ventricular Remodeling
As described elsewhere in this text, HF is characterized by progressive and deleterious LV remodeling. A large body of evidence implicates Ang II and aldosterone in the remodeling process, and ACEIs have been shown to prevent pathological hypertrophy, myocardial fibrosis, and progressive left ventricular dilation. These favorable effects of ACEIs were elegantly demonstrated in the rat coronary ligation model developed by Pfeffer et al. (37). In this model, MI was induced by occlusion of the left anterior descending (LAD) artery, which subsequently resulted in left ventricular systolic dysfunction (LVSD) and HF. Pfeffer et al. demonstrated that long-term ACE inhibition with captopril prevented or attenuated progressive LV dilation and deterioration of systolic function, and improved survival (38).
These initial animal studies paved the way for human studies investigating the effects of ACEI therapy on LV remodeling (Fig. 26-7). Pfeffer et al. performed a double-blind, placebo-controlled, randomized trial in patients with their first MI and LVEF <45% (39). After administration of captopril for 12 months, LV end-diastolic volume had increased by 21 ± 8 mL in patients randomized to placebo, compared to 10 ± 6 mL in patients randomized to the ACEI (p <0.02). The antiremodeling effects of captopril were most apparent in a subset of patients at exceptionally high risk due to persistent occlusion of the LAD artery. Thus, ACE inhibition with captopril appeared to prevent or attenuate the process of left ventricular remodeling which commonly occurs after MI. This beneficial effect provided the rationale for future trials of ACEI therapy on clinical endpoints in patients with HF.
Angiotensin-Converting Enzyme Inhibitors After Myocardial Infarction
Several well-designed randomized, controlled trials demonstrated the favorable effects of ACEIs when given prophylactically after MI. ACE inhibition appears to attenuate ventricular dilation, reduce hospitalizations for HF, prevent recurrent ischemic events, and increase survival.
The post-MI ACEI trials may be considered to fall into two categories: those studying the effects of ACE inhibition in patients with clinical HF and/or LVD, and those studying the short-term effects of ACEIs in unselected post-MI patients. The former category includes the Survival and Ventricular Enlargement trial (SAVE) (28), the Acute Infarction Ramipiril Efficacy Study (AIRE) (29), the Survival of Myocardial Infarction Long-term Evaluation study (SMILE) (40), and the Trandolapril Cardiac Evaluation study (TRACE) (41). These trials demonstrated impressive benefits of ACEI therapy, with risk reductions for all-cause mortality of 19%, 27%, 24%, and 22%, respectively.
The second category of post-MI trials involved the use of ACEIs for several weeks to 6 months in unselected patients. These studies, which include GISSI-3 (42), ISIS-4 (43), and the Chinese Cardiac Study (44), demonstrated mortality risk reductions of 12%, 7%, and 5%, respectively (Table 26-3). These landmark trials, which demonstrated the consistent, significant benefits of ACEIs, provided evidence for the routine use of ACEIs in all post-MI patients, regardless of HF symptoms or degree of left ventricular dysfunction. The one exception, CONSENSUS II (45), studied intravenous enalaprilat administered immediately after admission for acute MI, followed by oral enalapril, and demonstrated an adverse trend in mortality. This finding has been attributed to potentially deleterious effects of the early decline in blood pressure, especially in patients without evidence of HF. Thus, it appears that patients with poor left ventricular function or large areas of infarction appear to gain the most benefit as this is the group at highest risk for adverse left ventricular remodeling and future cardiovascular events, and that in these patients early and sustained ACE inhibition is warranted.
Angiotensin-Converting Enzyme Inhibitors in Asymptomatic Left Ventricular Dysfunction
The SOLVD-Prevention (46) study, which was performed in parallel with the SOLVD-Treatment study, enrolled patients with asymptomatic LVD. In many ways, SOLVD-Prevention can be viewed in the context of the post-MI studies in that most patients had prior MIs and were still susceptible to further remodeling and the development of clinical HF. In this study, administration of enalapril delayed the development of clinical HF (22.3 months versus 8.3 months in placebotreated patients) and significantly reduced the risk for HF hospitalizations. This was associated with less LV dilatation (47) in a population with serial measurements of LV
volumes. There was also a nonsignificant trend toward improved survival. Of note, after 10 years of follow-up those patients randomized to enalapril demonstrated a significant improvement in long-term survival (48). The SOLVD-Prevention study lent further support to the growing body of evidence that ACEIs are beneficial in all patients with reduced LV systolic function.
volumes. There was also a nonsignificant trend toward improved survival. Of note, after 10 years of follow-up those patients randomized to enalapril demonstrated a significant improvement in long-term survival (48). The SOLVD-Prevention study lent further support to the growing body of evidence that ACEIs are beneficial in all patients with reduced LV systolic function.
Table 26-2 Effects of Ace Inhibitors on Mortality in Patients with Chronic Heart Failure | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
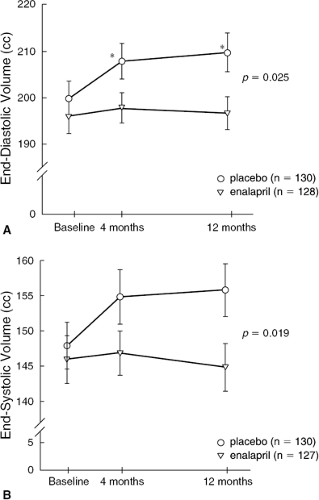 Figure 26-7 The echocardiographic substudy of SOLVD was designed to assess the effects of enalapril on changes in LV volumes and mass over a 1-year period. Changes in LV end-diastolic (A) and end-systolic (B) volumes are demonstrated. Enalapril significantly attenuated remodeling in this subset of the SOLVD patient population. (Reproduced from Greenberg B, Quinones MA, Koipillai C, et al. Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction. Results from the SOLVD echocardiography substudy. Circulation. 1995;91:2573–2581 , with permission.) |
Angiotensin-Converting Enzyme Inhibitors in Patients at High Risk for the Development of Left Ventricular Dysfunction
Given the consistent benefit derived from ACE inhibition in patients with HF, further research efforts addressed the utility of ACE blockade as primary prevention in patients deemed at to be high-risk for the development of LVSD. The first major trial to address this issue was the HOPE (Heart Outcomes Prevention Evaluation) (49) study, which enrolled patients with documented coronary, cerebrovascular, or peripheral vascular disease. HOPE randomized 9,541 patients to ramipril (10 mg per day) or placebo, and followed them for the development of HF, cardiovascular events, or stroke. This positive trial was terminated early due to the impressive 23% relative risk reduction for the development of HF in the ramipril-treated group (9.2% versus 11.7%, p = 0.002). The ramipril arm also had a 22% reduction in the primary endpoint of cardiac death, nonfatal MI, or stroke.
The HOPE study was followed by the EUROPA (European trial on Reduction Of cardiac events with Perindopril in stable coronary Artery disease) (50) trial, which also assessed the effects of ACE inhibition in patients with stable coronary heart disease and no known history of HF. In EUROPA, 13,665 patients with prior MI, history of coronary revascularization, a positive stress test, or angiographic coronary artery disease were randomized to perindopril or placebo for a mean of 4.2 years. Only 8% of patients assigned to perindopril, compared to 10% of placebo-treated patients, experienced the primary endpoint of cardiovascular death, MI, or cardiac arrest (RRR 20%, 95% CI 9%-29%; p = 0.0003). The results of EUROPA, combined with HOPE, led to the widespread use of ACEIs in patients known with coronary artery disease or other high-risk features, even in the absence of LVSD.
The recently completed PEACE (Prevention of Events with Angiotensin Converting Enzyme inhibition) (51) study, in which low-risk patients with known coronary artery disease and LVEF >40% were randomized to trandolapril or placebo, failed to reproduce the findings of HOPE and EUROPA. At 5 years follow-up, there was no significant difference in the combined primary endpoint of cardiovascular death, MI, or coronary revascularization between the two groups, although the point estimate for all-cause mortality was similar to that seen in HOPE and EUROPA (Fig. 26-8). The lack of significant benefit in the ACEI-treated patients is perhaps explained by the low-risk nature of the patients studied [average LVEF = 58% and blood pressure (BP) = 133/78 mm Hg] and the much more aggressive medical management that they received, compared to the earlier studies, with 90% treated with antiplatelet agents, 60% receiving beta-blockers, and 70% on lipid-lowering therapy. Thus, the additional benefit of ACE inhibition may be diminished in low-risk patients who are maximally treated with other anti-ischemic, anti-remodeling therapies. Nonetheless, ACEIs remain a mainstay of therapy for prevention of HF in high-risk patients and are usually given concurrently with other proven medications (Table 26-4).
Indications, Adverse Effects, and Contraindications for Angiotensin-Converting Enzyme Inhibitor Use
As previously reviewed, there is a large body of evidence supporting the routine use of ACEIs in patients with mild, moderate, and severe HF. ACEIs confer hemodynamic and
symptomatic improvement, prevent progression of LV dysfunction, and provide a strong mortality benefit for patients with HF due to LVSD, especially in patients with LVEF ≤35%. Patients with asymptomatic systolic dysfunction experience a reduction in the progression to clinical HF and, most likely, a modest improvement in long-term survival. There is strong evidence supporting the use of ACEIs in the setting of acute MI, particularly when clinical HF, systolic dysfunction, or large infarction is present, in order to improve survival, prevent the occurrence or worsening of HF, and to prevent recurrent cardiovascular events. Finally, ACEIs are also effective in preventing vascular events in other high-risk groups of patients.
symptomatic improvement, prevent progression of LV dysfunction, and provide a strong mortality benefit for patients with HF due to LVSD, especially in patients with LVEF ≤35%. Patients with asymptomatic systolic dysfunction experience a reduction in the progression to clinical HF and, most likely, a modest improvement in long-term survival. There is strong evidence supporting the use of ACEIs in the setting of acute MI, particularly when clinical HF, systolic dysfunction, or large infarction is present, in order to improve survival, prevent the occurrence or worsening of HF, and to prevent recurrent cardiovascular events. Finally, ACEIs are also effective in preventing vascular events in other high-risk groups of patients.
Table 26-3 Effects of Ace Inhibitors on Mortality in Patients with Post-Milv Dysfunction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


