Inflammatory Responses to Cardiopulmonary Bypass
Karsten Bartels
Mustafa Zakkar
The development of the heart-lung machine—cardiopulmonary bypass (CPB) in the 1950s—together with the development of cardiac catheterization and the use of heparin and protamine, made open-heart surgery possible and led to enormous improvement in patient longevity and quality of life (1). CPB imposes a complex set of nonphysiologic circumstances during which patients are subjected to severe physiologic alterations. With current anesthetic and surgical care paradigms, few serious adverse sequelae occur in most patients (2,3,4). Moreover, multiple recent large-scale clinical trials comparing off-pump coronary artery bypass grafting (CABG) surgery to conventional CPB (5,6,7,8) have demonstrated few meaningful differences, calling into question the clinical relevance of inflammation attributable to CPB alone (Table 13.1).
Inflammation is one of the oldest pathophysiologic concepts known to mankind—Galen and Celsus are credited with describing the pentad of calor (heat), rubor (redness), tumor (swelling), dolor (pain), and function laesa (altered function) over 2,000 years ago (9). Immunologic responses provide protection from a wide array of external pathogens, irritants, and endogenous substances. Historically, the immune system has been categorized into the innate and the adaptive (or acquired) immune system (10,11). The core feature of the adaptive immune system is its ability to establish memory from joint processing by lymphocytes and antigen-presenting cells (11). In case of reexposure to a previously encountered antigen, antibodies can then be rapidly generated. The innate immune system consists of hematopoietic cells such as neutrophils, eosinophils, mast cells, macrophages, and natural killer cells, as well as nonhematopoietic cells such as endo- and epithelial cells (10). In sterile inflammation, these can be activated by damage-associated molecular patterns (DAMPs), sterile particulates, and intracellular cytokines that are released from damaged cells (12). The innate immune response is then further mediated by soluble factors such as complement, cytokines/chemokines, and acute phase proteins. It should be noted that the innate and adaptive pathways of immunity are highly interrelated and that crosstalk is common.
THE SYSTEMIC INFLAMMATORY RESPONSE TO CARDIOPULMONARY BYPASS
Definition
At the outset, it is important to define what the systemic inflammatory response means both in general and in the specific context of CPB. Both clinical and experimental studies indicate that various infectious and noninfectious conditions induce a similar host response known as “systemic inflammatory response syndrome” (SIRS). This concept was advanced by the American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) consensus conference in 1992 (5,6,15,16). Two or more of the following clinical manifestations must be fulfilled for the diagnosis:
Body temperature higher than 38°C or lower than 36°C.
Heart rate more than 90 beats/min.
Respiratory rate more than 20/min or PaCO2 less than 32 mm Hg.
Leukocyte count more than 12,000 cells/mm3 or less than 4,000 cells/mm3, or the presence of more than 10% immature neutrophils.
The 2001 International Sepsis Conference pointed out the nonspecific nature of the SIRS criteria and further specified the variables that are likely to be affected by an infectious inflammatory stimulus (17). CPB can produce a “whole-body” noninfectious or “sterile” inflammatory response (18). Inflammation that occurs after CPB could well be described as an SIRS. However, discriminating the postoperative inflammatory component that derives from the surgical procedure versus the exposure to CPB proves challenging. Systemic inflammatory response after cardiopulmonary bypass (SIRAB) therefore includes inflammatory response from other components of cardiac surgery such as sternotomy or thoracotomy. Organ dysfunction associated with cardiac surgery can provoke the activation of a plethora of proinflammatory cascades adding to SIRAB (19). Hypotensive episodes during CPB can
lead to hypoxia and ischemia, which are strong proinflammatory activators and can initiate more inflammatory responses that can last long after the termination of bypass and manifest as a vasoplegic state (20). While most patients experience few clinically adverse sequelae from CPB, and only a minority develop severe hemodynamic changes or organ failure (21,22), length of CPB has been independently associated with morbidity and mortality after cardiac surgery (23).
lead to hypoxia and ischemia, which are strong proinflammatory activators and can initiate more inflammatory responses that can last long after the termination of bypass and manifest as a vasoplegic state (20). While most patients experience few clinically adverse sequelae from CPB, and only a minority develop severe hemodynamic changes or organ failure (21,22), length of CPB has been independently associated with morbidity and mortality after cardiac surgery (23).
TABLE 13.1. Summary of primary outcomes in recent clinical trials comparing offpump CABG surgery to conventional CABG using cardiopulmonary bypass | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
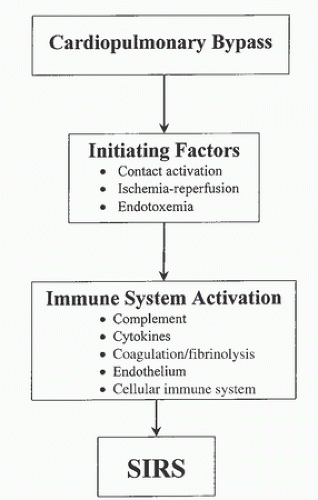 FIGURE 13.1. Schematic diagram of the sequence of events by which cardiopulmonary bypass (CPB) may lead to the development of systemic inflammatory response syndrome (SIRS). (From Warltier DC, Laffey JG, Boylan JF, et al. The systemic inflammatory response to cardiac surgery: implications for the anesthesiologist. Anesthesiology 2002;97(1):215-252. Copyright © 2002, American Society of Anesthesiology. Reproduced with permission from Wolters Kluwer Health.) |
Initiation of Systemic Inflammatory Response After Bypass
A number of injurious processes that impinge on both cellular and noncellular (humoral) elements of blood initiate SIRAB. These processes generate microemboli, disrupt hemostasis, and lead to a generalized whole-body inflammatory response. Most importantly, they set in motion a sequence of cytokine-mediated events that activate vascular endothelium, allowing further neutrophil-mediated inflammatory injury. The damaging effects of CPB are most commonly attributed to contact of blood with the foreign surface of the extracorporeal circuit. However, other factors such as altered arterial blood flow patterns, shear stress generated by blood pumps, cardiotomy suction devices, tissue ischemia and reperfusion, hypothermia, anemia, and heparin, may be equally important (24). This inflammatory response may continue long after CPB discontinuation, depending partly on its magnitude (25).
The repeated passage of blood through the nonphysiologic extracorporeal circuit initiates a “contact activation” response with deposition of fibrin and activation of contact protein
cascades upon exposure of the blood to the nonendothelialized surface of the CPB machine. Four proteins are involved in the contact activation cascades: coagulation factors XII and XI, prekallikrein, and high-molecular-weight kininogen (HMWK). This activation forms bradykinin and converts plasminogen into plasmin. This in turn initiates fibrinolysis and can trigger the classical complement cascade. Therefore, multiple inflammatory mediators are released upon exposure to the extracorporeal circuit (26). These responses initiate a sequence of chemokine-mediated events that activate vascular endothelium and potentiate neutrophil-mediated injury, which can disrupt hemostasis and create a generalized inflammatory response.
cascades upon exposure of the blood to the nonendothelialized surface of the CPB machine. Four proteins are involved in the contact activation cascades: coagulation factors XII and XI, prekallikrein, and high-molecular-weight kininogen (HMWK). This activation forms bradykinin and converts plasminogen into plasmin. This in turn initiates fibrinolysis and can trigger the classical complement cascade. Therefore, multiple inflammatory mediators are released upon exposure to the extracorporeal circuit (26). These responses initiate a sequence of chemokine-mediated events that activate vascular endothelium and potentiate neutrophil-mediated injury, which can disrupt hemostasis and create a generalized inflammatory response.
Cellular Components of Blood
Red Blood Cells
Tissue oxygenation depends upon continuous oxygen delivery, most of which is carried by the hemoglobin molecules in the red blood cells (RBCs). The extracorporeal circuit exposes RBCs to a challenging environment, thereby altering their integrity and function. Rheologic shear stress forces mechanically damage RBCs to reduce their deformability, which is important to maintaining normal RBC microcirculatory flow. As a further consequence of membrane distortion, RBCs are susceptible to the membrane attack complex (MAC) generated by the activation of complement (18), leading to hemoglobin leakage into the plasma. Free plasma hemoglobin may impair tissue function by increasing plasma oncotic pressure and viscosity. The ability of RBCs to aggregate also diminishes as a consequence of the dilution of blood by the CPB circuit priming solution. In addition to the obvious potential reduction in tissue oxygenation, this can trigger endothelial activation pathways (27).
Vascular Endothelium
Under resting conditions, vascular endothelium offers a relatively inert surface that regulates the passage of intravascular substrates to the extravascular space and ensures unhindered flow of cellular and serum components through the capillary network. Endothelial cells are extremely sensitive to the physiologic trespass imposed by CPB and surgical manipulation (and hypoxia, should it occur). Release of endogenous substances such as cytokines (IL-1b and tumor necrosis factor [TNF]-α), thrombin, C’5a, and lipopolysaccharide (endotoxin) activates endothelial cells to increase surface expression of E-selectin, endothelial leukocytes adhesion molecule (ELAM), intercellular adhesion molecule-1 (ICAM-1), and vascular adhesion molecule (VCAM), all of which mediate leukocyte recruitment (28,29,30) (Fig. 13.2).
Leukocytes
The recruitment of leukocytes from the circulation to an inflammatory site is a multistep process (Fig. 13.2) involving members of several adhesion receptor families (31,32). CPB activates vascular endothelium, leading to rapid expression of members of the selectin family (33). These are transmembrane molecules, expressed on the surface of leukocytes and activated endothelial cells. Selectins contain an N-terminal extracellular domain with structural homology to calcium-dependent lectins, followed by a domain homologous to epidermal growth factor, and two to nine consensus repeats (CR) similar to sequences found in complement regulatory proteins. Each of these adhesion receptors is inserted through a hydrophobic transmembrane domain and possesses a short cytoplasmic tail. During inflammation, bloodstream leukocytes attach to endothelium via selectins, thereby impairing microcirculatory flow, potentially culminating in transient standstill from leukocyte adhesive “plugs” along the endothelium. The selectin family includes P-, E-, and L-selectin. P-selectin is largely responsible for the rolling phase of the leukocyte adhesion cascade. Transmigration, the next step, is mediated by adhesion molecule upregulation, including platelet endothelial cell adhesion molecule (PECAM)-1, CD99, ICAM-1, CD11a/CD18, very late antigen (VLA)-4, and endothelium-secreting chemoattractants (34).
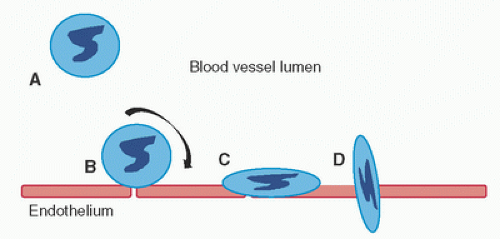 FIGURE 13.2. Adherence of circulating neutrophils (A) is triggered by activation of the endothelial cells, which causes the neutrophils to roll along the endothelium (B), a process mediated by selectin interaction with carbohydrate ligands. Activation of the neutrophils (e.g., by interleukin [IL]-8, C5a, or platelet activating factor [PAF]) results in activation of integrin molecules (e.g., by leukocyte function antigen [LFA]-1) on the neutrophil surface. This results in firm adhesion of the neutrophils to activated endothelium (C) through receptors belonging to the Ig family (e.g., intercellular adhesion molecules [ICAMs]). Activated adherent neutrophils extravasate through the endothelium into the tissue interstitial space (D). |
Neutrophils
Neutrophils are key mediators of the systemic inflammatory response; their recruitment, activation, and cytotoxic capability are essential to warding off infection. Neutrophil activation through interaction with activated vascular endothelium may be responsible for much of the clinical sequelae of SIRAB. Activation of neutrophils during CPB is shown by the loss of L-selectin and the upregulation of CD11b/CD18 (Mac-1). Increased production of reactive oxygen intermediate (35) and neutrophil-derived elastase and similar molecules have also been reported (35,36). Studies in complement-deficient dogs suggest that at least some of the neutrophil activation seen in
CPB is due to complement activation (37,38). In addition, the release of cytotoxic products by activated cells can cause direct cellular damage. These cytotoxins include both preformed agents present in neutrophil granules and newly synthesized molecules. These novel substances include leukotrienes (36) and reactive oxygen intermediates (35). This synthesis can be readily detected by a dramatic increase in cellular oxygen consumption. These species are largely responsible for the so-called ischemia-reperfusion injury. Ischemic injury occurs when the blood supply to a tissue is impaired or suboptimal, which may occur with CPB. The paradox, however, is that a more severe tissue injury occurs when blood flow is restored upon reperfusion.
CPB is due to complement activation (37,38). In addition, the release of cytotoxic products by activated cells can cause direct cellular damage. These cytotoxins include both preformed agents present in neutrophil granules and newly synthesized molecules. These novel substances include leukotrienes (36) and reactive oxygen intermediates (35). This synthesis can be readily detected by a dramatic increase in cellular oxygen consumption. These species are largely responsible for the so-called ischemia-reperfusion injury. Ischemic injury occurs when the blood supply to a tissue is impaired or suboptimal, which may occur with CPB. The paradox, however, is that a more severe tissue injury occurs when blood flow is restored upon reperfusion.
Monocytes
Peripheral monocytes are also involved in the systemic inflammatory process. They possess migratory, chemotactic, pinocytic, and phagocytic activities, as well as receptors for IgG and C’3b. Upon migration to tissue, they undergo further differentiation to become tissue macrophages, which participate in both specific and nonspecific immune pathways. Recruitment of these cells occurs as early as 1 hour following insult (39) and is thought to be mediated mainly by monocyte chemoattractant protein-1 (MCP-1), although other factors such as C’5a play an important role. Upon activation, they play a pivotal role in inflammation, serving as effector cells secreting cytokines (40). Monocytes can exhibit both pro- and anti-inflammatory properties, depending on the signals they receive, and it has been suggested that these cells have the capacity to switch from one state to the other to participate in both the induction and the resolution of inflammation (39).
Platelets
CPB is associated with a transient deficit in platelet function and number, which can impair postoperative hemostasis (41). The normal platelet adheres to a damaged endothelial cell or the subendothelial layer. The multimeric form of von Willebrand factor forms a bridge between endothelium and platelet at the platelet glycoprotein (GpIb) receptor site. The platelet then undergoes a conformational change to expose different glycoproteins, including the GpIIb/IIIa complex, which can in turn bind to fibrinogen. Fibrinogen is an essential cofactor in platelet adhesion and in platelet-to-platelet binding during irreversible aggregation. The protein complex thrombospondin stabilizes this platelet aggregate. Concurrent thromboxane A2 release produces local vasoconstriction and further platelet aggregation.
Numerous CPB-related factors contribute to platelet changes. These include physical factors (e.g., hypothermia and shear forces), exposure to artificial surfaces (42), drugs, and endogenous chemicals (43,44). Hemodilution contributes to initial thrombocytopenia (45), but mechanical disruption, adhesion to the extracorporeal circuit, and sequestration in organs explains a disproportionate drop (beyond hemodilution alone) in platelet count, which can be 30% to 50% below baseline (46). The response of platelets to CPB is complex and multifactorial, including rapid consumption of platelets during bypass (45), decreased reactivity to known agonists (44), increase in the concentration of the a-granule compounds in plasma (47), and an increase in the stable metabolite of thromboxane A2 (thromboxane B2) released from aggregating platelets (48). Morphologic changes, including spherical appearance and the development of pseudopods, also occur during CPB (49). Prolonged bleeding time after CPB correlates with duration of bypass; however, its time course and precise mechanism remain unclear (50).
Platelets activated during CPB form conjugates among themselves and between platelets and leukocytes. Activated platelets express P-selectin, which contributes to leukocyte conjugate formation by binding PSGL-1 (51




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
