Fig. 21.1
Hypothesized mechanisms of acute venous thrombosis post activation of the venous endothelium. The activation of tissue factor, P-selectin and E-selectin, platelets, inflammatory cells, and microparticles lead to thrombus formation and amplification (From Stanley et al. [12] with permission)
It is widely known that, under normal conditions, cellular blood components interact with the vessel wall in order to promote normal vascular repair.
The effect of the thrombus on the cellular components of the vein wall is not well defined; however, post thrombosis, the vein wall increases in stiffness and thickness and loses compliance [13]. The non-injured vein wall at rest is characterized by a continuous endothelium, a thin and compliant structure with scattered vascular smooth muscle cells (VSMCs) (Fig. 21.2). In contrast, the post-thrombotic vein wall is thickened, with loss of endothelium (early) and disrupted matrix and proliferative VSMC. The longer an occlusive or partially occlusive thrombus is in contact with the vein wall, the greater the inflammatory response and damage to the vein wall as defined in a recent review by DeRoo et al. [13] (Fig. 21.2).
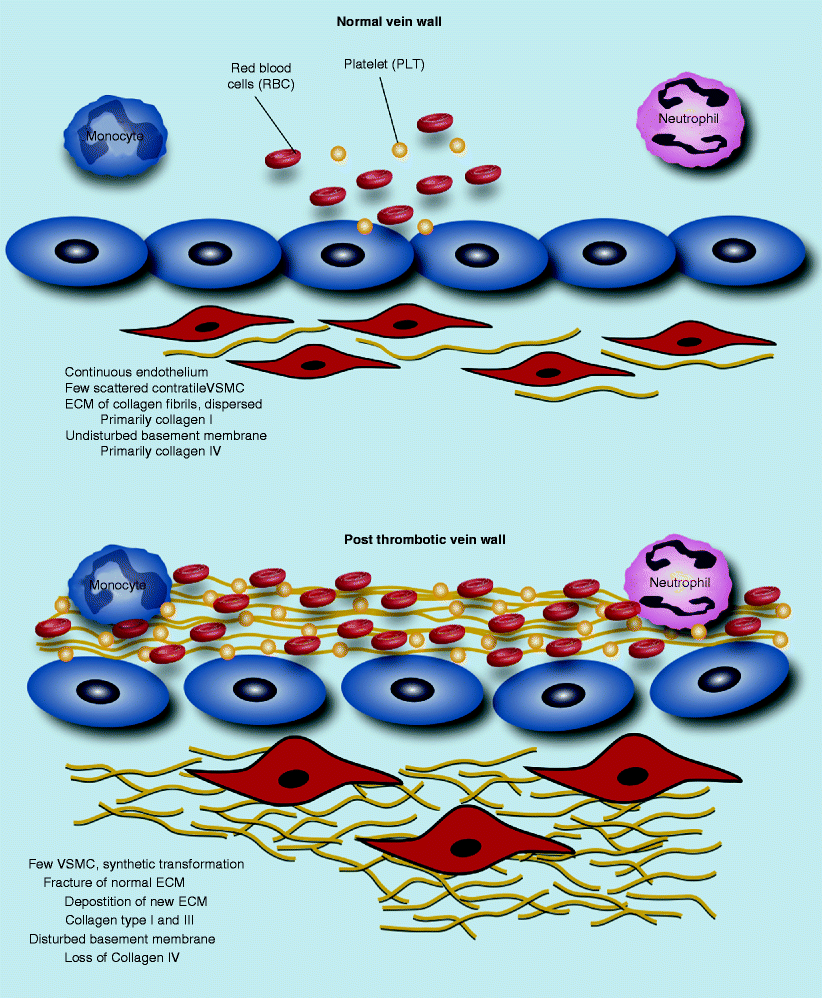
Fig. 21.2
Vein wall matrix changes post venous thrombosis (Modified from DeRoo et al. [13] with permission)
Inflammation and Thrombogenesis
There is evidence to suggest that inflammation and thrombosis interact and have mechanisms in common. Selectins are glycoproteins that are expressed by leukocytes, activated endothelial cells, and platelets. The role of selectins is to mediate the initial adhesion interactions of leukocytes stimulated by physiological changes in blood flow at sites of vascular endothelial injury. Three selectins have currently been identified: P-selectin, E-selectin, and L-selectin [14]. P- and E-selectins are cell adhesion molecules with critical roles in thrombogenesis. Animal studies using rat and mouse venous thrombosis models demonstrated upregulation of P-selectin and E-selectin in the vein wall 6 h and 6 days after thrombus induction, respectively [14] (Fig. 21.1). P-selectin is involved in leukocyte rolling and adhesion, an early inflammatory mechanism that facilitates leukocyte transmigration. The P-selectin receptor, P-selectin glycoprotein ligand 1 (PSGL-1), is a glycoprotein expressed on the surface of leukocytes and platelets that plays a critical role in the recruitment of leukocytes and platelets into inflamed tissue. The interaction of PSGL-1 with P-selectin (the endothelial cell P-selectin: PSGL-1-leukocyte complex) promotes rolling and adhesion of leukocytes and platelets, which ultimately result in increased vein wall cell infiltration [15] (Fig. 21.1).
A study by Wagner et al. showed that the increase in the number of P-selectin molecules present on the endothelial cell surface is due to the release from the Weibel-Palade body (WPB). WPBs are the endothelial-specific storage organelle for regulated secretion of von Willebrand factor (vWF) and P-selectin onto its membrane [15]. Thus, the exocytosis of WPB initiates a rapid translocation of P-selectin to the endothelial surface, resulting in augmented endothelial adhesiveness for leukocytes and platelets. Soluble P-selectin (sP-sel) is released from activated platelets and endothelial cells, and levels rise significantly during pathologic conditions. The function of sP-sel has been shown to be an endogenous activator of coagulation via generation of plasma microparticles in addition to its ability to bind to PSGL-1, leading to leukocyte recruitment and rolling [16].
Circulating cell-derived microparticles (MP) contribute to the coagulation and amplification of thrombosis (Fig. 21.1). They are present in the blood of healthy individuals. These MPs are small vesicles (less than 1 μm) consisting of a plasma membrane surrounding a small amount of cytoplasm with cell-specific surface molecules [17]. Endothelial cells, leukocytes, and platelets have a very well-structured plasma membrane characterized by a controlled transverse lipid distribution termed “rafts.” The activation of these cells promotes a general membrane content redistribution during which rafts concentrate in areas of the cell that will ultimately derive in MP. Therefore, the MP membrane is rich in lipid rafts. These MPs concentrate tissue factor (TF) in caveolae where it is stored with the tissue factor pathway inhibitor (TFPI). The fusion of MP with activated platelets promotes thrombus formation in a TF-dependent manner. In addition to TF, the expression of prothrombinase activity on the membrane [18] and PSGL-1 [11] are involved in the procoagulant activity of MPs (Fig. 21.1).
During inflammation, activation of endothelial cells upregulates surface expression of P-selectin, leading to formation of the endothelial cell-P-selectin: PSGL-1-leukocyte complexes. These complexes stimulate the production of MPs from leukocytes, particularly monocytes, along with platelets and endothelial cells. In addition, the accumulation of leukocyte markers expressed on the surface of MPs in the growing thrombus is mediated by the P-selectin-PSGL-1 complex [11]. Therefore, MP concentration increases dramatically at the area of vein wall injury and inflammation [19]. MPs also possess a phosphatidylserine-rich anionic surface capable of assembling complexes of the coagulation cascade. Another molecule expressed on the MP membrane surface is PSGL-1, which then can bind to upregulated P-selectin on platelet surfaces in the thrombus. There is even evidence that macrophage-1 (Mac-1) on leukocyte-derived MPs can allow interactions between MPs and inactivated platelets using glycoprotein 1b alpha (GP1ba), resulting in further platelet activation with P-selectin upregulation [20, 21]. All these events occurring in the area of thrombus formation lead to thrombus amplification. The increase of circulating MPs with the onset of inflammation adds to the proposed mechanisms linking vein wall inflammation and thrombogenesis (Fig. 21.1).
Circulating inhibitory mechanisms regulating the process of thrombogenesis include anti-thrombin III (ATIII), protein C, protein S, TFPI, and glycocalyx-associated glycosaminoglycans, such as heparin sulfate, endothelial protein C receptor, and thrombomodulin (TM), which facilitates inhibitory activity of thrombin by anti-thrombin III and APC by TM.
Anti-thrombin III, a plasma glycoprotein synthesized in the liver, is a serine protease inhibitor (SERPIN) structurally related to other plasma protease inhibitors, such as alpha 1-antichymotrypsin, alpha 2-antiplasmin, and heparin cofactor II. Anti-thrombin III acts as a pseudo-substrate for the inhibition of the intrinsic pathway (factors IIa [thrombin], IXa, Xa, XIa, XIIa) and the extrinsic pathway (factor VII), kallikrein, and plasmin. Other targets of anti-thrombin III include trypsin and the C1s subunit, which are involved in the classical complement pathway. The anti-thrombin III plasma half-life is 60–70 h, while the thrombin-antithrombin (TAT) complex is cleared by the liver, and its inhibitory activity is increased by heparin [1].
Protein C, a vitamin K-dependent plasma glycoprotein, is synthesized as a single chain and cleaved prior to secretion by the liver. Plasma protein C is a two-chain molecule, consisting of a light and heavy chain. Its half-life is 6–7 h. Once protein C binds to its receptor, endothelial protein C receptor, it is cleaved (activated) by the thrombin-thrombomodulin complex on the endothelial surface, resulting in activated protein C (APC). In the presence of calcium and protein S, APC inactivates factor Va and factor VIIIa of the “protein C anticoagulant pathway.”
Protein S, a vitamin K-dependent plasma glycoprotein, is synthesized by the liver, the endothelial cells, and the megakaryocytes. Protein S is a cofactor of APC in the protein C anticoagulant pathway. In addition, protein S exhibits APC-independent anticoagulant activity by binding to factors Va, VIIIa and Xa [1]. In serum, protein S is found in two forms: free (active) and protein bound (inactive). Almost 70 % of circulating protein S is bound to a complement protein (C4b-binding protein). The remaining 30 % circulates as “free protein S,” which has a half-life of 96 h. It is the free protein S that acts as a cofactor for protein C [1].
TFPI is a single-chain plasma polypeptide that inhibits factor Xa and TF-factor VIIa complex catalytic activity. Plasma only contains 20–30 % of the intravascular minor fraction of TFPI, and it is largely bound to lipoproteins. The majority of TFPI (60–70 %) is normally bound to the vascular endothelium. This pool of TFPI is released into circulation after heparin injection [1].
Prostacyclin and nitric oxide (NO) are secreted by endothelial cells. These compounds synergistically contribute to vessel homeostasis by reducing vascular smooth muscle cells tone and growth, platelet aggregation, leukocyte adhesion to endothelium, and susceptibility to thrombosis. Interestingly, Osanai et al. demonstrated that vessel homeostasis might be maintained through an increase in prostacyclin production in vascular endothelial cells when NO synthesis is impaired [22].
We recently used our mouse inferior vena cava (IVC) ligation model to generate venous thrombosis in order to study the role of interleukin-6 (IL-6) in thrombus development. Specifically, an antibody (anti-IL-6) was administered to mice pre-thrombus induction that leads to in vivo neutralization [23]. We found that IL-6 had an acute and a chronic effect. During the acute phase (2 days post-thrombosis), C-C motif chemokine ligand 2 or CCL-2 (the mouse equivalent to human monocyte chemotactic protein-1 [MCP-1]) was significantly decreased in the group where circulating IL-6 was blocked. This was demonstrated by the gene expression and protein level [23]. The decrease in CCL-2 caused a reduction in the number of monocytes recruited into the injured area at an intermediate time point (6 days post thrombosis), indicating a dynamic and linked process. At the chronic time point (14 days post thrombosis), a statistically significant decrease in fibrosis was observed in the group treated with anti-IL6 vs. control [23]. This was demonstrated using several methods, including Masson’s stain quantification of fibrosis. In summary, we have shown a relationship between IL-6/CCL-2/monocyte recruitment and fibrosis for the first time in the context of VT, exhibiting a potential pathway that started early on in venous thrombosis (VT) and continued to have an impact on chronic VT as well [23]. This is an important step in understanding the link between inflammation and thrombosis in the context of VT and indeed a recent insight into the molecular and cellular contributions to venous thrombosis.
The role of neutrophil extracellular traps (NETs) in acute inflammation was recently described. In addition to their phagocytic and bactericidal functions, activated neutrophils have been shown to release chromatin deoxyribonucleic acid (DNA) and histone containing granular antimicrobial proteins, which form extracellular matrices or traps [24]. The mechanisms regarding neutrophil cell death and the formation of NETs appear to be separate from apoptosis and necrosis and appear to rely upon the formation of intracellular reactive oxygen species [25]. NETs have been shown to kill bacteria while forming a microbial containment barrier [26, 27]. In addition, NETS have been reported to form during sepsis and in inflammatory non-infectious disease states such as small-vessel vasculitis [28, 29].
It was demonstrated that NETs were associated with DVT when using an experimental baboon model of occlusive balloon-induced iliac deep vein thrombosis [30]. Plasma DNA samples collected at baseline were significantly lower than plasma DNA samples collected at 2 and 6 days post-DVT (p < 0.01) [30]. Furthermore, the rise in plasma DNA kinetics was similar to our previous findings of plasma D-dimers in the same experimental model [31]. In addition to plasma DNA, histologic samples of iliac veins (6 days post-DVT) and controls were stained using an antibody directed toward DNA histone complexes (nuclear origin) and evaluated for the presence of DNA nuclei and extracellular DNA or NETs. Affected vessels demonstrated punctate staining of nuclear DNA and diffuse staining of extracellular DNA compared to controls. Furthermore, immunocolocalization of VWF strings were dispersed within the DNA core and between the DNA core and the vessel wall [30]. These are the first reported results demonstrating that markers of NETs were present in both the plasma and thrombus in experimental DVT. These results provide early evidence regarding the interactions and roles of NETs (DNA and histones), vWF, and platelet binding in venous thrombosis.
Plasminogen Activators and Thrombolysis
Venous thrombosis is a dynamic process with thrombus formation (thrombogenesis) and dissolution (thrombolysis) occurring almost simultaneously under normal conditions in a healthy individual [1]. Thrombolysis depends on multiple physiological processes, including fibrinolysis. In response to thrombus formation, natural anticoagulants such as protein C and protein S have roles in thrombolysis. Similarly, circulating plasminogen is activated to plasmin, which is the main fibrinolytic enzyme [11]. Plasmin’s main substrates include fibrin, fibrinogen, and other coagulation factors. In addition, plasmin interferes with vWF-mediated platelet adhesion by proteolysis of GpIb receptor complex [32].
Plasminogen activators are serine proteases that activate plasminogen by the proteolytic cleavage of a single arginine-valine peptide bond. Plasminogen activator inhibitor type 1 (PAI-1) is the primary inhibitor of the plasminogen activators, both tissue type plasminogen activator (t-PA) and urokinase type plasminogen activator (u-PA), and hence fibrinolysis [11]. PAI-1 is produced by the endothelium but is also secreted in an active form by the liver and adipose tissue. Increased PAI-1 levels are found in various disease states such as cancer, obesity and metabolic syndrome. Thus, the increased occurrence of thrombosis in patients with these conditions has been suggested to be associated with elevated PAI-1 levels. PAI-1 elevation appears to synergize with factor V Leiden genetic abnormalities [11]. It is possible that elevated PAI-1 levels could suppress fibrinolysis and increase thrombosis, hence increasing the clinical manifestations of DVT. However, studies on the role of elevated levels of PAI-1 to venous thrombosis have been contradictory [33].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


