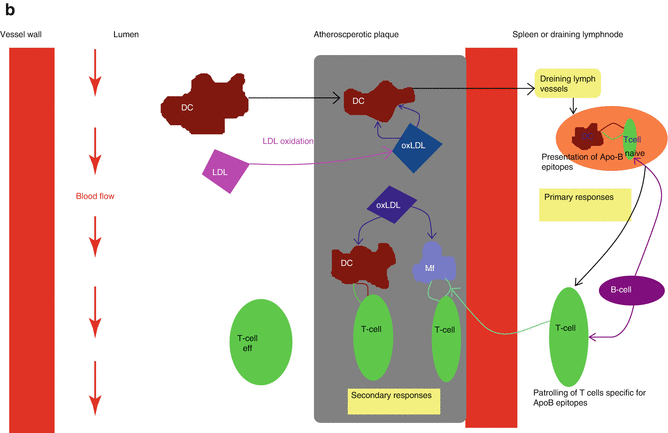
Fig. 13.1
The activation of innate and adaptive immune response in atherosclerosis. (a) The innate immune response. Modified LDL particles taken-up by scavenger receptors may activate the inflammasome, leading to IL-1β secretion. Adjunctive signals come from modified LDL activation of TLRs. NF-kB, IRF and AP-1 transcription factors are then activated and a wide variety of proinflammatory molecules is produced. (b) The adaptive immune response. Activated DCs present naïve T cells with antigens such as ApoB100. The latter develop into effector T cells that enter the bloodstream. When recruited within atherosclerotic plaques, they are reactivated by antigens presented by local macrophages, DCs or B cells (Adapted by permission from Macmillan Publishers Ltd: Hansson and Henmarsson [6]. Copyright 2012) Abbreviations: ApoB apolipoprotein beta, AP-1 activated protein 1, DC dendritic cell, IL interleukin, IRF insulin related factor, IP inositol triphosphate, LTB4 leukotrien beta, Mf macrophage, MCP monocyte chemoattractant protein, MyD88 myeloid differentiation primary response 88, NLPR3 Nod-like protein 3, NFkB nuclear factor kappa beta, oxLDL oxidized LDL, TLR toll-like receptor, TNF tumor necrosis factor
13.2.3 The Inflammasome and the Role of NLPR-3 Inflammasome in Atherosclerosis
Inflammasomes are a group of cytosolic multiprotein complexes that recognize inflammation-inducing stimuli including pathogen associated molecular patterns (PAMPs) and DAMPs and control the production of pro-inflammatory cytokines such as IL-1β and IL-18, through the activation of caspase-1. They are also involved in pyroptosis, a form of cell death associated with tissue repair. Under normal circumstances, inflammasome activity needs to be tightly regulated to avoid excessive deleterious cytokine production. Indeed, alternative splicing, subcellular location and trafficking of its components, direct sequestration of protein complexes and inducible down-regulation by inflammatory cytokines and CD4+ cells, are some of the mechanisms that protect the cell from the dire consequences of inflammasome overactivation. Inflammasome mediated processes are important for microbial infections as well as metabolic processes and mucosal defense. Inflammasome activation has diverse effects on different cell lineages, namely the production of acute phase proteins by the liver, prostaglandin E2 release by brain, and chemoattraction of neutrophils and platelets within bone tissue (Fig. 13.2a).
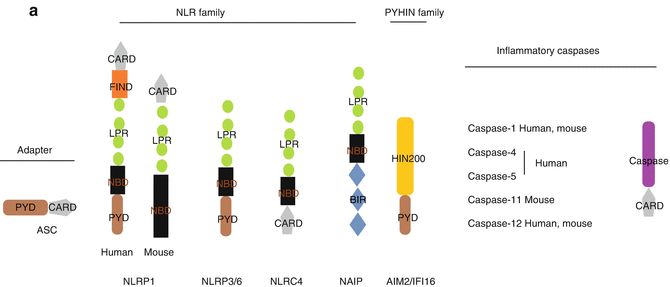
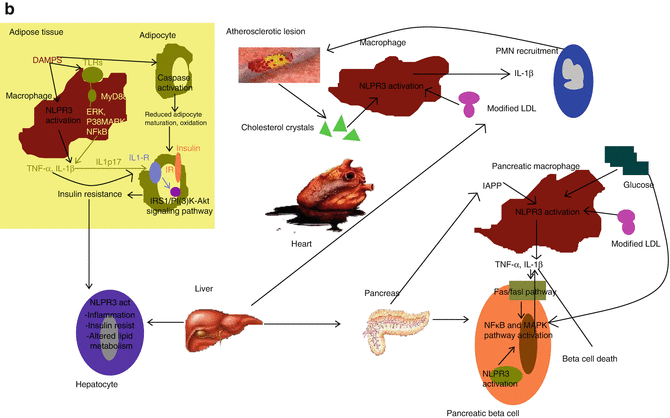
Fig. 13.2
The inflammasome. (a) The organization of the inflammasome. Core components of the inflammasome belong to two families, namely NOD-like receptor (NLR) and the PYHIN (pyrin and HIN 200 domain) protein family. The NLR family (NLRP1, NLRP2, NLRP3, NLRP6 and NLRC4 and NLRP12) contain a nucleotide binding domain (NBD), carboxy-terminal leucine rich repeat (LRR) and either a PYD or a caspase activation domain (CARD) or both. The PYHIN family members AIM2 and IFI16 have a PYD and a HIN 200 domain commited to ligand binding. (b) The role of the inflammasome in metabolic syndrome. DAMPS activate NLPR3 inflammasome in various tissues in obese patients. Palmitate and ceramides activate NLPR3 inflammasome in activated macrophages of adipose tissue. Caspase-1 activation regulates adipocyte differentiation and leads to fatty acid oxygenation. Within the pancreas, both resident macrophages and beta cells secrete IL-1β upon NLPR3 recruitment by IAPP and ROS. Beta cell death and impaired insulin production ensue (Adapted by permission from Macmillan Publishers Ltd: Strowig et al. [2]. Copyright 2012). Abbreviations: BIR baculoviral inhibition of apoptosis repeat domain, DAMP damage associated molecular pattern, ERK extracellular signal related kinase, FIINF domain with function to find, IAPP islet amyloid polypeptide, IL interleukin, IL-1R interleukin-1 receptor, IP(3)-Akt insulin receptor protein kinase B, IRS insulin receptor substrate, MyD88 myeloid differentiation primary response 88, NLPR3 nod-like protein 3, PMN neutrophil, p38MAPK protein 38 mitogen activated protein kinase, TLR toll-like receptor, TNF tumor necrosis factor
The most widely studied is the NLPR-3 inflammasome, normally activated during viral and parasitic infections. The NLPR-3 inflammasome also assembles in response to a diversity of signals. Phagosome- or mitochondria- derived ROS, lysosome aggregates comprising cathepsins B and L and cellular potassium efflux act synergistically to promote NLPR-3 activation. Aberrant activation of NLPR-3 by non-infectious agents is implicated in a wide variety of diseases characterized by sterile INFL such as gout and pseudogout, silica/asbestos related diseases, Alzheimer’s disease and in metabolic diseases, but also in ATH.
The contribution of inflammasome to the pathogenesis of ATH may be both direct – through activation by cholesterol crystals- and indirect. In the first case, cholesterol crystals within macrophages activate NLPR-3 through phagolysosome destabilization [7]. In vivo priming of NLPR-3 inflammasome implicates TLR-4/6 and MyD88 dependent pathways and various scavenger receptors; mice deficient in any of these components exhibit less ATH [8]. LDL-receptor deficient mice -which are prone to atherosclerosis- are resistant to disease when genetically modified not to express NLPR-3 or IL-1β. However this finding was not confirmed when APOE-deficient mice –another ATH murine model- were knocked out for NLPR-3 and fed with a very high fat diet [9]. This discrepancy may imply the existence of potent environmental factors that may alter the course of the disease, the deterministic effect of knocking out genes preferentially at early stages rather than when ATH has been established. This data suggest the presence of yet undefined redundant mechanisms that may account for the activation of the inflammatory cascade independently of the NLPR3 assembly [4].
The indirect effects of inflammasome activation on ATH have emerged from its role in metabolic syndrome, obesity and diabetes (Fig. 13.2b). Insulin resistance, impaired pancreatic beta-cell function, fatty liver disease and ATH are thought to be components of the “metabolic syndrome” and are associated with increased IL-1β and IL-18 production which is the hallmark of NLPR-3 activation. Within adipose tissue, increased levels of palmitate and other saturated free fatty acids reduce the activation of AMP-activated kinase (AMPK), a key regulator of lipid metabolism. This in turn leads to defective mitochondrial autophagy, culminating in the accumulation of dysfunctional mitochondria which release enhanced levels of ROS and DNA into the cytosol. Both these molecules promote the activation of NLPR-3 inflammasome and the release of IL-1β which hampers the function of both, insulin-target cells and pancreatic beta cells. More specifically, IL-1β inhibits the engagement of insulin receptor (IR) by the insulin receptor substrate 1 (IRS1) and the phosphoinositie 3 kinase [PI(3)K-] signaling pathway, which leads to insulin resistance. In the pancreas, islet amyloid polypeptide (IAPP), which is secreted together with insulin, may act synergistically with glucose to induce NLPR-3 mediated macrophage derived IL-1β release, which causes beta cell dysfunction and death through the activation of Fas-FasL pathway and MAPK/ΝF-κB signaling cascade. Caspase-1 activation is also capable of altering hepatic tissue functions. Enhanced NLPR-3 activation has been demonstrated in patients with non alcoholic steatohepatitis (NASH). A role of the NLPR-3 inflammasome in shaping the adaptive immune response has also been postulated to contribute to the above-mentioned process. In this context, NLPR-3 driven, aberrant accumulation of lymphocytes into adipose tissue, and commitment to the TH17 lineage, may further enhance insulin resistance and promote obesity. Along these lines, exercise and calorie restriction were shown to reduce NLPR-3 and IL-1β expression in obese individuals and mice genetically modified not to express NLPR-3 were protected from obesity-induced inflammasome activation in their adipose tissue [10]. Furthermore, inflammasome contributes to the progression of type 2 diabetes, via thioredoxin-interacting protein (TXNIP)-, an ROS sensitive inducer of NLPR3 that senses islet amyloid polypeptide (IAPP)-, which may trigger further inflammasome activation within atherosclerotic lesions and ultimately atherosclerosis progression and plaque destabilization. Of note, glyburide, which is an insulin secretagogue used for the treatment of diabetes, suppresses NLPR-3 mediated IL-1β release which points towards the potential anti-inflammatory actions of the drug as being of major importance for its antidiabetic effects [11].
TLR “priming” of inflammatory cells can provide indispensable adjunct signals for effective inflammasome activation. The significance of TLRs to inflammasome activation is underscored by the fact that under normal circumstances, inflammasome components such as NLPR-3, apoptosis associated speck like protein containing a CARD (ASC) and caspase-1 are expressed at extremely low or even undetectable levels. The necessary induction of the expression of inflammasome components as well as pro-IL-1β and pro-IL-18 seems to be accomplished through TLR activation upon their interaction with DAMPs –such as HSP-60 and HMGB1- or even dietary-derived ligands which come under the term “nutrition associated molecular patterns”, (NAMPs). Oxidized LDL ligates to a receptor complex engaging a TLR-4/6 heterodimer and the receptor CD36 to induce IL-1β expression [9]. Minimally modified LDL (mmLDL) also induces TLR-4 dependent Syk- and Nox-2 mediated macrophage derived ROS production, which is another essential signal for inflammasome activation, NLPR-3 up-regulation and IL-1β release.
Another aspect of the combined TLR/NLPR-3 activation in atherosclerosis is related to its unique effect on cellular survival signals. In this context, scavenger receptor class A (SR-A) and TLR-4 combined signaling within endoplasmic reticulum stressed macrophages induces proapoptotic signals potentially related to enhanced plaque necrosis and destabilization, which is not the case upon single TLR stimulation. In this case oxLDL, oxidized phospholipids, lipoproteins and saturated fatty acids may act as ligands for the doubly committed pathway. The potentially protective effects of omega-3 poly-unsaturated fatty acids through GPR120 mediated attenuation of inflammasome activation remain to be further established [12].
Genetic variation of inflammasome related loci may also alter the expression profile of several cardiovascular disease related factors such as blood pressure, acute phase proteins and fibrinogen. These data originate from genome wide association and other genetic studies but the precise role of genetic determinants of inflammasome expression on cardiovascular disease burden needs to be better elucidated.
13.3 The Endothelium in Inflammation and Atherosclerosis
The endothelium is the principal regulator of vascular wall homeostasis. Normally, endothelial cells preserve a relaxed vascular tone combined with a low level of oxidative stress. This is accomplished through the release of mediators such as nitric oxide (NO), prostacyclin (PGI2) and endothelin (ET) and the tight control of local angiotensin II activity. In addition, the endothelium actively regulates vascular permeability to plasma constituents, leukocyte and platelet adhesion and aggregation. A wide variety of conditions are capable of altering this balanced endothelial regulation and divert the endothelium from a “homeostatic” to “a non-adaptive” state, which comes under the term “endothelial dysfunction”. Pro-inflammatory stimuli such as diet rich in saturated fat, obesity, insulin resistance and hypertension activate the endothelium by increasing levels of circulating cytokines such as IL-1 and TNF-α. This up-regulates endothelial expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), VCAM-1, P-selectin and V-selectin and facilitates the capture, rolling and firm tethering of leukocytes to the endothelial surface. Adhered monocytes then transmigrate into the intima in a monocyte chemoattractant protein 1 (MCP-1) mediated process that also involves macrophage colony stimulating factor (MCSF) and initiates or propagates local INFL. Studies have shown that patients with cardiovascular risk factors, but no overt cardiovascular disease have endothelial dysfunction, which is per se an independent predictor of future cardiac events.
13.3.1 LDL and HDL Cholesterol
There is a well documented association between atherogenic lipoproteins (such as some forms of LDL, post-prandial chylomicron remnants and fasting triglyceride-rich particles) and endothelium-dependent responses. Hypercholesterolemia and high levels of LDL are inversely related to endothelium dependent vasodilation whereas anti-atherogenic high density lipoproteins (HDL), apart from mediating reverse cholesterol transport, also seem to modulate endothelial function in a beneficial manner. Oxidized LDL plays a pivotal role in endothelial activation. Accumulated modified LDL molecules retained within the arterial intima activate endothelial cells which up-regulate the expression of adhesion molecules such as VCAM-1 and RANTES, a phenomenon that occurs predominantly at sites undergoing increased hemodynamic stress. This leads to the subsequent recruitment of leukocytes and monocytes, and the propagation of local inflammation. Post-prandial hypertriglyceridemia is also associated with increased levels of TNF-α, IL-6, soluble ICAM-1 (sICAM-1) and sVCAM-1, which imply a pro-inflammatory effect for triglycerides in endothelial dysfunction and atherogenesis.
Plasma levels of HDL are strongly and inversely correlated with ATH. Their atheroprotective effects have been attributed to several mechanisms including reverse cholesterol transport, antithrombotic/anti-inflammatory properties and importantly, their effects on endothelial repair.
HDLs have been shown to prevent LDL oxidation and inhibit MCP-1 mediated macrophage transmigration into the intima. They also downregulate the expression of adhesion molecules and they inhibit the sphingosine kinase, extracellular signal regulated kinase (ERK) and NF-kB signaling cascades of pro-inflammatory cytokine production. Vacular smooth muscle cell proliferation depends on the inflammatory chemokines chemokine ligand 2 (CCL2), CCL5 and CX3CL1. Through NF-kB/Akt and pERK inhibition, HDLs reduce SMC chemokine expression and proliferation contributing to the stabilization of atherosclerotic plaques. It is also postulated that HDL may protect against oxidative damage. Thus, some of its cardioprotective effects may rely on its ability to increase endothelial e-NO synthase (e-NOS) activity and NO production (Table 13.1). HDLs from humans with systemic inflammatory disorders such as systemic lupus erythematosus or rheumatoid arthritis may be incapable of preventing LDL oxidation, suggesting that HDL composition and function may be altered during inflammation and that it can shift from an anti-inflammatory to a pro-inflammatory status under specific conditions (see below). Recent information on the atheroprotective actions of HDLs may justify their use as therapeutic targets for the prevention of atherosclerotic lesion formation and the regression and/or stabilization of the established atherosclerotic plaque [13].
Table 13.1
Atheroprotective and antiiflammatory actions of HDL
Reverse cholesterol transport |
Inhibition of LDL oxidation |
Down-regulation of MCP-1, inhibition of macrophage transmigration to the intima |
Alterations in signaling cascades of proiflammatory cytokines (NF-kB, ΕRK) |
Reduction of endothelial SMC proliferation |
Enhancement of e-NOS activity and NO production |
13.4 Interactions Between Inflammation and Lipid Metabolism
Inflammation and dyslipidemia are well established risk factors for the development of ATH. Lipid lowering drugs exert their atheroprotective effects, through the decrease of atherogenic lipids and other “off target”, anti-inflammatory actions. On the other hand, anti-inflammatory drugs, may partially restore an abnormal lipid profile in subjects with systemic INFL. High through-put technologies –which are described later- in animal models and in human tissues, have unraveled the existence of an interplay between environmental factors, lipid metabolism and INFL, which is of crucial importance for the pathogenesis of ATH. Free cholesterol and TG, modified LDL and free fatty acids (FFAs) can all alter the “inflammatory status” of an organism through a variety of mechanisms (Fig. 13.3).
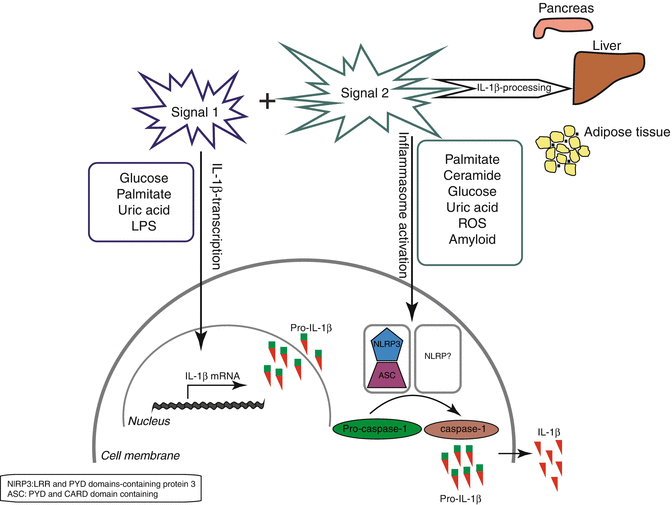
Fig. 13.3
Inflammasome activation in metabolic syndrome*. Overview of the metabolic triggers that may act as primary or secondary signals in inflammasome NLPR3 activation and IL-1β production. This signaling cascade occurs in various tissues such as pancreas, liver, adipose tissue contributing to tissue dysfunction and insulin resistance (Reprinted from Stienstra et al. [1]. Copyright 2012, with permission from Elsevier). Abbreviations: ASC apoptosis associated speck like protein containing a CARD, IL interleukin, IL-1R interleukin-1 receptor, LPS lipopolysacharide, NLPR3 nod-like protein 3, ROS reactive oxygen species
Cholesterol feeding elevates markers of INFL such as CRP. Obese patients also exhibit chronic low INFL as evidenced by increased levels of CRP and cytokines such as TNF-α and IL-6. “Metabolic inflammation” is believed to be primarily induced by metabolic endotoxemia, that is, an increased translocation of lipopolysacharide (LPS or endotoxin) through the gut, upon high fat diet. The additive effect of cholesterol and/or TG induced up-regulation of NF-kB mediated cytokine production by several tissues including adipose tissue, liver and vasculature are also important. In addition to high fat diet per se, modified lipoproteins may also activate hepatic macrophages and vascular endothelial cells –through the recruitment of the inflammasome- and downregulate the function of TREGS. Importantly, adverse hemodynamic conditions may well activate sterol regulatory element binding protein-2 (SREBP2) –a lipogenic transcriptor factor which is aberrantly regulated in NASH- and induce NLPR-3 inflammasome in endothelial cells within atheroprone vascular areas, thus acting concomitantly with hyperlipidemia to increase ATH [14].
Nuclear lipid sensors comprise a specific group of transcription factors that upon recognition of intracellular lipids, regulate the expression of genes implicated in the inflammatory cascade. Peroxisome proliferator activated receptors (PPARs) are fatty acid sensors capable of modifying macrophage response and leukocyte recruitment. Liver X receptors (LXRs) are activated by oxysterols in response to increased intracellular cholesterol levels and exert their anti-inflammatory effects through the activation or reverse cholesterol transport, reduction of inflammatory gene expression and inhibition of T-cell proliferation through control of cholesterol content/membrane synthesis of T-cells. Finally, the farsenoid receptors (FXRs) are activated by endogenous bile acids and expressed at high levels in the liver and the intestine. Their activation regulates bile acid, lipid and glucose metabolism, concurrently leading to FXR binding with inflammatory transcriptional co-repressors or silencing mediators, which ultimately translates into modulation of INFL.
Importantly, lipid metabolism can be modified through epigenetic processes induced by the microbiome, that is the assembly of integrally associated microorganisms colonizing individuals. It has been demonstrated that alterations in the microbial population of the gut can induce metabolic changes favoring the development of metabolic syndrome, obesity and diabetes. Increased plasma levels of trimethylamine-N-oxide (TMAO), an intestinal microbiota-dependent metabolite of choline which is also induced by the farsenoid X receptor, were associated with cardiovascular events in a dose-dependent way in 4,000 individuals studied with elective coronary angiography [15]. The promotion of thiol-dependent oxidant stress, macrophage activation, LDL oxidation and methionine methylation are some of the mechanisms explaining the atherogenic actions of TMAO. Importantly, these complex atherothrombogenic host-microbiota interactions could be potentially modified through dietary changes, the use of probiotics and pharmacological interventions that can suppress the synthesis of TMAO.
Although the effects of lipids on inflammatory pathways have been extensively studied, the role of inflammation on lipid metabolism is addressed only by few studies. Acute inflammation in the context of infection results in increases in TG levels with reduction of both LDL and HDL cholesterol and concomitant increase in oxygenation of LDL particles which facilitate host defense. However, chronic INFL may induce detrimental, atherogenic effects. The most relevant studies come from patients with systemic inflammatory and/or autoimmune conditions, who display an abnormal lipid metabolism and increased incidence of CAD.
13.5 Accelerated Atherosclerosis in Autoimmune Inflammation
Autoimmune diseases (AIDs) are a heterogeneous group of disorders characterized by humoral induced cell-mediated immune responses against self-constituents. The combined effect of complex predisposing genetic factors, immune conditions and environmental triggering events is important to AID manifestations. Among AIDs, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), antiphospholipid syndrome (APS) and Sjogren syndrome (SS) display augmented ATH linked to an increased cardiovascular morbidity and mortality that cannot be attributed to the classical predisposing risk factors for CAD. There is a growing body of evidence in favor of a causative link between chronic autoimmune inflammation and accelerated atherosclerosis in patients with AIDs.
Cardiovascular disease in SLE patients has been reported with an overall incidence of 6–10 % in various cohorts. Strikingly, women with SLE display a 5- to 9- fold increased risk for CAD compared with their non-diseased counterparts, and premenopausal status does not seem to confer to them adequate cardiovascular protection. Endothelial impairment is one of the key issues in the pathogenesis of ATH in SLE patients. This is induced by the integrated effects of proinflammatory cytokines –such as TNF-α, IFN-γ and IL-10-, the direct toxic actions of cytotoxic T-cells and the autoreactive B-cell mediated complement-dependent attack of the endothelium, all of which synergistically act to destroy its integrity and allow the propagation of inflammation and atherogenesis. Importantly, circulating immune complexes and anti-endothelial autoantibodies are capable of inducing a pro-inflammatory and pro-adhesion endothelial cell phenotype. Defective functions of TREGS –that do not sufficiently suppress autoreactive T-cells- and enhanced TH17 activity promoting INFL and tissue destruction are also implicated in SLE-related ATH. Interestingly, patients with SLE display increased antibody titers to oxLDLs, and these correlate with disease activity scores and complement activation. Furthermore, antibodies to HDL and apoA1 are also elevated in patients with active SLE. Of note, these correlate inversely with the activity of paraoxonase which normally confers to HDL its anti-oxidant properties [16]. Chronic INFL may cause a decrease in HDL concentration and impair its functions through several alternative mechanisms; these include the enhancement of endothelial lipase and soluble phospholipase A2 activities as well as the replacement of apoA-1 in HDL with serum amyloid A. Modified HDL molecules have been identified as activators of protein kinase Akt, which in turn regulates apoptotic signals on endothelial, smooth muscle cells and pancreatic beta cells, and drives the production of adhesion molecules and NO within the endothelium. Thus, in the context of chronic INFL, HDL may lose its atheroprotective properties and become pro-atherogenic [17]. On the basis of these observations, it can be concluded that, SLE may confer increased cardiovascular risk by instigating both, endothelial damage and alterations in lipid metabolism.
RA is characterized by synovial inflammation and hyperplasia, autoantibody (anti-CCP and RF) production, cartilage tissue and bone destruction and systemic manifestations such as pulmonary or cardiovascular disease. Increased expression of adhesion molecules, pro-inflammatory cytokines and matrix metalloproteases all contribute to joint erosion and atherosclerosis in rheumatoid patients. Tumor necrosis factor alpha (TNF-α) is a key cytokine to RA pathogenesis. This cytokine has a pivotal role in the disruption of microvascular and macrovascular circulation in patients and animal models of the disease. Through NF-kB dependent and independent pathways, it is capable of inducing the expression of various other inflammatory cytokines, chemokines and ROS that contribute to endothelium apoptosis, vascular oxidative stress, impaired NO bioavailability, neovascularization and vascular INFL. It also impairs insulin sensitivity by interfacing with insulin receptor signaling at the level of insulin receptor substrates through activation of kinase enzymes that phosphorylate serine of threonine residues, thus regulating cell proliferation, apoptosis and differentiation, namely serine kinases . Importantly, environmental factors such as diet, smoking and aging have a strong impact on TNF-α production. In addition, some dietary supplements and exercise act favorably on vascular function, since they diminish TNF-α production and inhibit ΤΝF-α mediated signaling [18].
On the basis of the importance of TNF-α for endothelial dysfunction, several clinical trials have investigated the use of the three currently available TNF-α inhibitors (infliximab, etanercept and adalimumab) in vascular disorders that accompany RA and other autoimmune conditions. A growing body of evidence -mainly derived from observational databases and registries- suggests that anti-TNF biologic agents as well as methotrexate, can reduce the risk of future cardiovascular events in patients with RA. Chronic treatment with either of the above-mentioned agents improves endothelial function in RA patients. Anti-TNF-α therapy ameliorates insulin resistance, beta-cell function and insulin signaling in individuals with active RA [19]. In addition to TNF-α inhibitors, various statins (atorvastatin and simvastatin) and the ACE-inhibitor quinapril have been found to decrease circulating levels of ΤΝF-α, which parallels the improvement of endothelial function in patients with type 2 diabetes, congestive heart failure, RA or hyperlipidemia [20, 21]. The role of TNF-α inhibition in CAD and type 2 diabetes remains to be further investigated on the basis of randomized controlled trials.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


