, Cyro Alves de Brito3 and Alberto José da Silva Duarte1, 4, 2, 5
(1)
Clinical Laboratory, Hospital do Coração, São Paulo, Brazil
(2)
Central Laboratory of Hospital das Clínicas, Faculty of Medicine, University of São Paulo, São Paulo, Brazil
(3)
Center of Immunology, Adolfo Lutz Institute, São Paulo, Brazil
(4)
Faculty of Medicine, University of São Paulo, São Paulo, Brazil
(5)
Laboratory of Medical Investigation Unit 56, Faculty of Medicine, University of São Paulo, São Paulo, Brazil
Abstract
Cardiovascular diseases (CVDs) are the leading cause of death worldwide. The World Health Organization has estimated that around 17.3 million people died from CVD in 2008, representing 30 % of all global deaths. Of these deaths, an estimated 7.3 million were due to coronary heart disease and 6.2 million to stroke [1].
Introduction
Cardiovascular diseases (CVDs) are the leading cause of death worldwide. The World Health Organization has estimated that around 17.3 million people died from CVDs in 2008, representing 30 % of all global deaths. Of these deaths, an estimated 7.3 million were due to coronary heart disease and 6.2 million to stroke [1].
The majority of the CVDs are induced by atherosclerosis, which is associated with a chronic inflammatory process in which the immune system has an important role. The natural evolution of this process is the formation of a thrombus that can cause a partial or complete blockage of the blood flow, inducing damage to the organ, such as the heart or brain.
Because of the blood flow blockage in a coronary artery, the ischemic environment results in myocardial infarction and the death of cardiomyocytes. The established disorder attracts substances out of the blood and retains them. These substances, such as cytokines and chemokines, are produced during the inflammatory response to ischemic injury, mediating various reactions and attracting leukocytes to the lesion.
Despite the clinical therapeutic advances for CVDs, surgery is still widely used to treat them, and it has been reported that major general surgery causes profound alterations in immunity. After an initial proinflammatory phase associated with innate immunity, an immunosuppressive response mediated mainly by cells of the adaptive immune system takes place. Therefore, surgical stress inhibits innate immunity from the time of incision until about the first postoperative day. This is a crucial period with greater susceptibility to bacterial infections. In generaly, this susceptibility may be caused by the absence of recovery of innate immunity and lead to postoperative complications. The goal of further clinical studies must be to establish the immunomodulating property of the individual, which is very important for controlling innate immunity and inflammation in each patient.
For understanding the alterations established in the postoperative period, we first describe how the inflammation, innate immunity, and adaptive immune response take place and sequentially understand which modifications are imposed by the surgery. Finally, we describe some trials attempting to modulate immunity to inhibit the immunosuppression generated by the perioperative period.
Inflammation
Inflammation is the body’s reply to injury or irritation, a protective attempt to both remove the stimulant and initiate the healing process. This response involves both innate and adaptive immunity, as well as diverse molecular and cellular processes (e.g., chemotaxis, phagocytosis, mitosis, and cell differentiation) by the affected tissues.
In the past, inflammation was associated with infections and the immune system. However, recent evidence suggests that a broader range of disorders have inflammation markers. These include cancer, acute cerebrovascular stroke, Alzheimer’s disease, chronic arterial and venous disease, myocardial ischemia, and systemic arterial hypertension [2].
The various forms of inflammation differ according to their location in the tissue, the organ where they occur, and the nature and severity of the tissue injury (e.g., mechanical injury, infectious agent, chemical injury, burn injury, radiation, tissue injury due to lack of organ perfusion, and oxygen supply) (Fig. 3.1a). In general, the inflammatory cascade includes several microvascular reactions, such as elevated permeability in microvessels, attachment of circulating cells to the vessels in the vicinity of the injury site, migration of several cell types, cell apoptosis, and growth of new tissue and blood vessels.
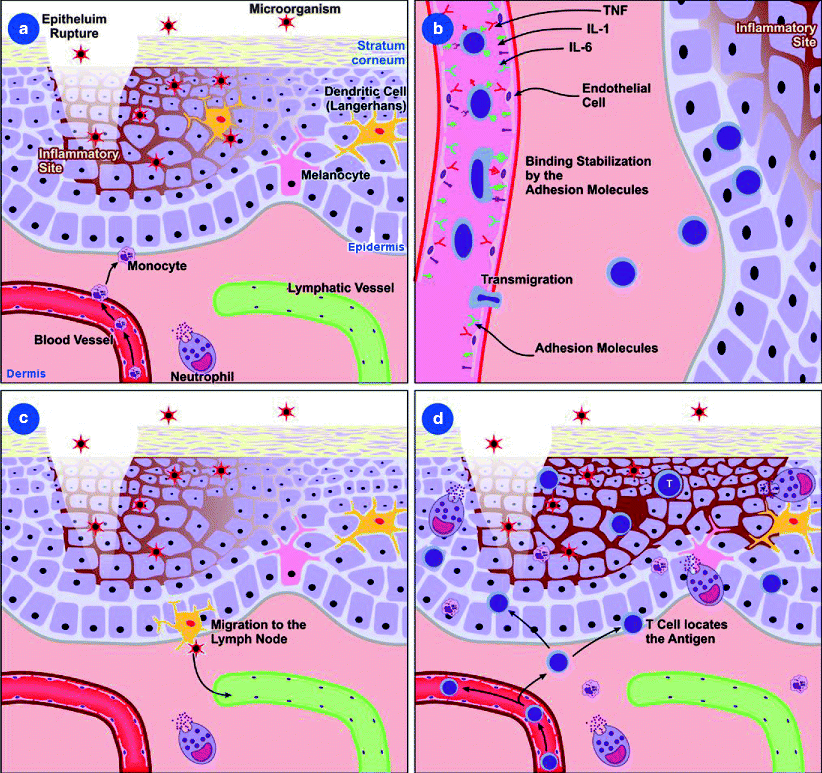
Fig. 3.1
Mechanisms involved in cell migration. (a) Inflammatory mediators, such as cytokines and chemokines, are produced during the inflammatory response to the pathogen or tissue proteins resulting from the cell damage. This response aims to isolate and destroy the pathogen and regenerate the injured tissue. (b) Expression of adhesion molecules facilitates the attachment of circulating cells to the endothelium and to tissue structures. The alteration in the blood flow allows the leukocytes in the blood to approach the endothelium, starting with a process of “rolling” by the interaction between the adhesion molecules expressed by the endothelial cells and the leukocyte molecules. At this time, the chemokines will influence the cell migration pattern. (c) Antigen-presenting cells migrate from the tissue to lymphoid organs where the adaptive immune response begins. (d) Activated lymphocytes leave the lymphatic circulation and migrate to the inflammation site
An important form of cell activation in the microcirculation is the expression of membrane adhesion molecules that facilitate the attachment of circulating cells to the endothelium and to tissue structures over which they migrate. The alteration in the blood flow allows the leukocytes in the blood to approach the endothelium, starting with a process of “rolling” by the interaction between the adhesion molecules expressed by the endothelial cells (E-selectin, expressed by the activated endothelial cell, and P-selectin, which has constitutive expression) and leukocyte molecules (L-selectins) (Fig. 3.1b). After this, the action of leukotriene B4, platelet activation factor, and interleukin (IL)-8 triggers a conformational change of the integrins expressed in the leukocytes [leukocyte function-associated (LFA)-1 or CD11a/18 and Mac-1 or CD11b/18] enabling a firm interaction with the intercellular adhesion molecule (ICAM)-1 and ICAM-2 molecules expressed by the endothelial cells. The interaction of the leukocytes with platelet endothelial cell adhesion molecules allows its passage to the tissue through diapedesis and migration to the inflammatory site. At this time, the chemokines will influence the cell migration pattern, IL-8 and CXCL7 being important to neutrophil migration, CCL2, CCL3, and CCL4 to monocyte migration, and CCL11 to eosinophil migration [3, 4].
Inflammatory mediators, such as cytokines and chemokines, are produced during the inflammatory response to the pathogen or tissue proteins resulting from the cell damage. This response aims to isolate and destroy the pathogen and regenerate the injured tissue (Fig. 3.1c, d). It starts with the contraction or dilation of the arteriole muscle wall and the dilation of the venules by contraction of the actin and myosin filaments of the endothelial cells. This mechanism is due to the action of the mediators produced by local mast cells and macrophages. Some of these mediators are histamine, lipid mediators (leukotrienes- B4, C4, and D4, prostaglandins, and platelet activation factor), and neuropeptides [substance p and calcitonin gene-related peptide]. Parallel to this event, there is the activation of the blood coagulation cascade (production of chemotactic peptides), fibrinolytic system, complement activation, and kinin system (production of bradykinin) [3, 4].
The activation of the cells involved in the innate immune response induces the secretion of various cytokines, such as IL-1, IL-6, IL-8, IL-12, IL-18, and TNF-α. IL-1 acts in the vascular endothelium, increasing leukocyte adhesion, and in the bone marrow, increasing leukocyte production. In synergy with IL-18, IL-1 triggers the intracellular signalization with the activation via nuclear factor κB (NF-κB ), which induces the production of many cytokines and chemokines [3, 4]. IL-1 and TNF-α bind to the thermoregulatory receptors in the hypothalamus, causing fever. TNF-α increases the vascular permeability of the endothelium, allowing the accumulation of immunoglobulins and complement proteins in the injured tissue. IL-6 is responsible for the stimulation and differentiation of the B lymphocytes and together with IL-1 and TNF-α acts in the liver, promoting the synthesis of the acute phase proteins, such as C-reactive protein (CRP). IL-8 is important in the cell migration to the damaged tissue, especially neutrophils, and IL-12, which is responsible for the activation of the natural killer cells (NK) [2].
Innate Immunity
The first line of defense of the organism against infections is composed by its natural barriers, which are formed by the skin and mucosal epithelial surface, the corporal fluids, and the normal bacterial flora (Fig. 3.2). The loss of integrity in any of these barriers, with or without infection, initiates the innate immune response. Other elements that compose the innate immunity include the complement system, inflammatory mediators, chemokines, cytokines, pathogen-associated molecular pattern recognition receptors, antimicrobial peptides, and some populations of the immune cells.
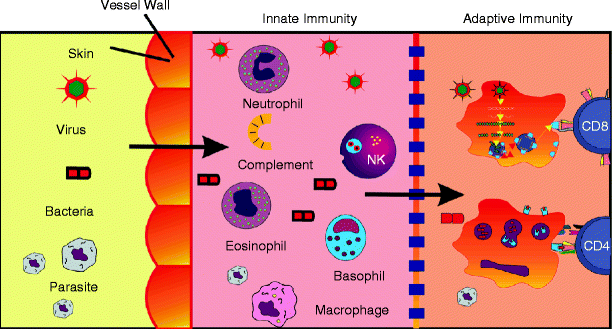
Fig. 3.2
Defense barriers against infections. The first line of defense is composed by the natural barriers, which are formed by the skin and mucosal epithelial surface, the corporal fluids, and the normal bacterial flora. The loss of integrity in any of these barriers, with or without infection, initiates the innate immunity. Next, the adaptive immune response is initiated, which is characterized by specificity and memory
The innate immune response depends on cells such as phagocytes, NK cells, NKT lymphocytes, γ:δ T cells, and the B-1 lymphocytes capable of directly recognizing antigens. The phagocytes comprehend the neutrophils and monocytes/macrophages, which recognize the pathogens opsonized by antibodies and C3b or by the CD14 and CD204 receptors. In phagocytosis antigens are internalized, forming a membrane vesicle named the phagosome, which joins with a pre-formed vesicle (lysosome) rich in substances that act in the elimination of the pathogen. This substances are produced primarily by the process of oxidative respiration [lysosomal oxidase, nicotinamide adenine dinucleotide phosphate (NADPH), and other enzymes] where several cytotoxic products derived from oxygen are formed, such as the superoxide anion (O2-), hydrogen peroxide (H2O2), free oxygen (O2), hydroxyl radical (OH), and hypochlorous acid (HOCl). Nitrogen oxides (nitric oxide-NO), antimicrobial peptides (cationic), lysozyme, acid hydrolases, and competitors, such as lactoferrin, an iron chelator, are also produced. The macrophages are also responsible for secretion of IL-12, a cytokine important to the cellular activation.
The NK cells can potentially secrete IFN-γ, which is vital to the control of infection by intracellular pathogens, such as the Herpes virus, Leishmania, and Listeria monocytogenes, before the activation of the antigen-specific T lymphocytes. Other cytokines involved in the innate response are interferon type α and β (IFN-α and IFN-β), which have the capacity to inhibit viral replication, activate NK cells, and increase the expression of major histocompatibility complex (MHC-1) molecules in the infected cells, increasing their susceptibility to attack by cytotoxic T cells.
Innate Receptors
As a result of evolutionary progress, all multicellular organisms have developed mechanisms to recognize invading organisms by specific receptors to structures common to the majority of pathogens. These molecular patterns associated with pathogens are called pathogen-associated molecular patterns (PAMPs). They are highly conserved products associated with several microorganisms, such as the bacterial outer membrane, lipopolysaccharides (LPS), glycopeptides (PGN), lipotechoic acid (LTA), cytosine-phosphate-guanine unmethylated motifs (CpG), double-stranded RNA from RNA viruses, and mannans from fungal walls [3].
However, molecular patterns associated with tissue damage are called damage/danger-associated molecular patterns (DAMPs) [5]. In theory, every single molecule that normally resides within the cells is a potential DAMP when released after cell death. In fact, the systemic and/or local DAMPS, such as heat shock proteins (HSP), surfactant protein A, and protein of the high mobility group 1 (HMGB1), are elevated in inflammatory and autoimmune disorders, including CVDs [6]. These molecules have gained much attention in DAMP-mediated activation of the innate immune system and subsequent inflammation after cardiac ischemia. The mechanisms by which DAMPs activate and control leukocyte behavior and whether DAMPs can be used as therapeutic targets in order to modulate detrimental inflammatory responses remain unclear.
PAMPs and DAMPs activate innate immunity through some receptors, contributing to the inflammatory process even in the absence of pathogens. The molecules and receptors capable of interacting with those molecules are called pattern-recognition receptors/molecules (PRRs/PRMs). PRRs are responsible for the cell/antigen interaction in the inflammatory site, collaborating in phagocytosis, cytokines secretion, and intracellular signalization. PRMs are soluble, secreted proteins, such as surfactant protein (SP)-A, SP-D, mannose binding protein (MBL), ficolins, and CRP.
Two categories of PRRs are recognized: (1) scavenger receptors, which mediate the capture, uptake, and presentation of antigen, and (2) Toll-like receptors (TLR), which lead the activation of proinflammatory pathways.
Toll-like receptors (TLRs) constitute a family of host defense receptors, which are expressed in different cell types, especially in immune system cells, but also in keratinocytes and epithelial cells, among others (Fig. 3.3). To date, 13 TLRs have been identified in mammals, including 10 in humans and 12 in mice [7]. Among the TLR ligands are microbial compounds, such as peptideoglycan (TLR 1, 2, 3, 6), viral double-stranded RNA (TLR 3), lipopolysaccharide (TLR 4), flagellin (TLR 5), single-stranded RNA (TLR 7, 8), bacterial and viral DNA containing non-methylated cytosine-guanine dinucleotide (CpG) (TLR 9) [8], and uropathogenic E. coli and T. gondii profilin (TLR 11 in mice) [9].
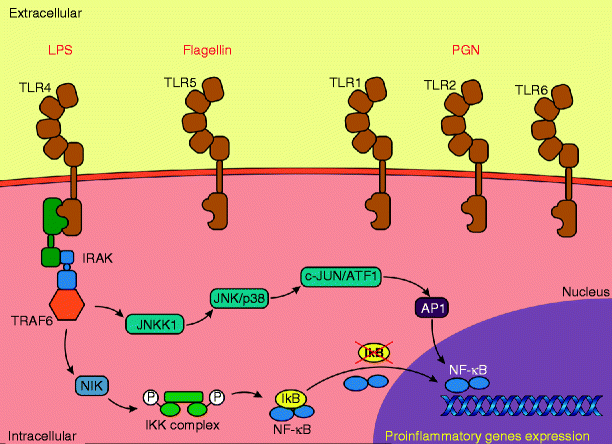
Fig. 3.3
Toll-like receptors (TLRs) constitute a family of innate immunity receptors. The intracellular events that occur during the activation of TLR are mediated by myeloid differentiation factor 88 (MyD88) or by the molecule TIR domain-containing adapter-inducing IFN-β (TRIF). TLR activation promotes activation of nuclear factor kappa B (NF-κB), which leads to production of proinflammatory cytokines
The intracellular events that occur during the activation of TLR are mediated by myeloid differentiation factor 88 (MyD88), except in the activation of TLR3, which is mediated by the molecule TRIF (TIR domain-containing adapter inducing IFN-β). The activation of TLR4 is mediated by both MyD88 and TRIF [10]. MyD88-dependent pathway promotes early activation of nuclear factor kappa B (NF-κB), which leads to production of proinflammatory cytokines, particularly TNF-α. Furthermore, the MyD88-independent pathway, mediated by TRIF, promotes activation of the late phase of NF-κB and interferon regulatory factor (IRF), inducing the synthesis of IFN-β and the expression of genes induced by type 1 IFN. The activation of NF-κB is well recognized as a hallmark of innate immunity activation after cardiac ischemia. As the signaling cascade triggered by TLRs also involves NF-κB, a new paradigm of noninfectious activation of the innate immune system following cardiac ischemia can be postulated.
In an experimental model, a rapid activation of Irak-1, an important kinase in the response via TLR, has been shown after transient ischemia. This activation is caused by increased levels of HSP60 binding to TLR4 in triggering the inflammatory process in cardiac tissue [11]. It was also observed that treatment of cardiomyocytes with HSP60 leads to the activation of caspase-3 and -8, suggesting the activation of apoptotic pathways. Indeed, HSP60 induces apoptosis in rat cardiomyocytes in part by binding to TLR4 [12]. Moreover, apoptosis induced by myocardial ischemia/reperfusion injury is significantly attenuated in mice deficient in TLR4 or treated with anti-HSP60 [11].
Adaptive Immunity
The main characteristics of the adaptive immune response are specificity and memory. Unlike the innate immune responses, adaptive immune responses are not initiated in the inflammatory site, but in the peripheral lymphoid tissues. It occurs after antigen-presenting cells (APCs), such as macrophages and dendritic cells, migrate from the inflamed tissue to the nearest lymphoid tissue, carrying antigen particles that are presented to T lymphocytes (Fig. 3.4).
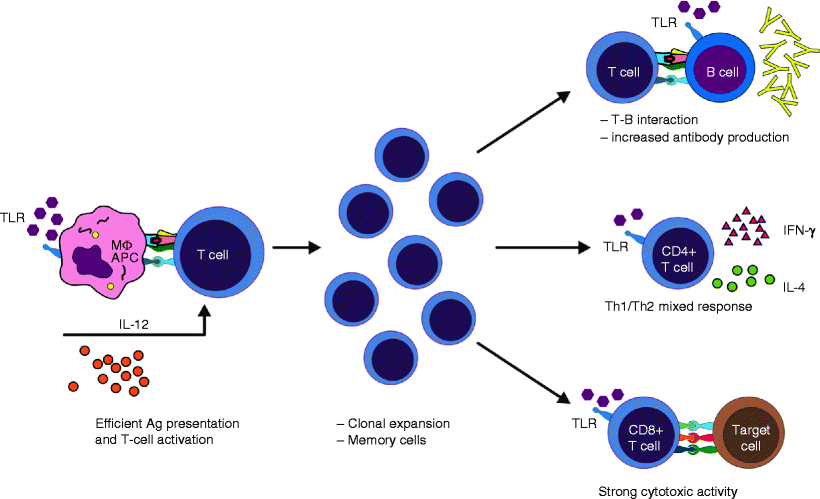
Fig. 3.4
Linking innate and adaptive immunity. Toll-like receptor agonists, such as DAMPs, increase expression of MHC and costimulatory molecules in APCs, causing proper activation of T cells, which promotes T cell clonal expansion and generation of memory cells. Immune adjuvants improve the interaction between T and B cells, secretion of Th1/Th2 cytokines, and antibody production, and they increase CTL activity in newborns (Adapted from de Brito et al. [13])
Unlike B cells, T lymphocytes recognize antigen only when it is associated with major histocompatibility complex (MHC) molecules expressed on the cell surface. The MHC molecules are glycoproteins encoded by highly polymorphic genes, also called HLA (human leukocyte antigen) genes in humans. There are two types of MHC molecules related to antigen presentation, the class I and class II, which differ in structure and expression pattern in different cell types. MHC-I molecules are composed of two polypeptide chains: a largest α chain, consisting of three domains, non-covalently linked to a smaller chain named β2 microglobulin. MHC-I is expressed on all nucleated cells of the body and associated with the peptides present in the cytosol, such as molecules of cytosolic pathogens, especially viruses, to present them to CD8+ T cells [3]. Class II MHC molecules, on the other hand, are expressed on antigen-presenting cells (APCs) and activated T lymphocytes. They consist of a non-covalent complex of two chains, α and β, each with two domains, which cross the membrane. These molecules bind to peptides derived from the degradation of antigens in vesicular compartments of macrophages, dendritic cells, and B lymphocytes, which will be presented to CD4+ T cells.
The binding of the complex peptide:MHC to a specific TCR is the first event necessary for activation of T lymphocytes. However, for the occurrence of an efficient activation of the immune response the involvement of accessory molecules is required. The binding of LFA-1 molecules, CD2 and ICAM-3, expressed by T lymphocytes, and ICAM-1 and 2, LFA-3, and dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) by APCs allows T lymphocytes to come into contact with large numbers of MHC molecules on the APC surface [4].
Another group of molecules involved in cell activation is the group of co-stimulatory molecules. In addition to antigen recognition via the TCR, a “second signal” or co-stimulatory signal is necessary for cell activation. The absence of co-stimulatory molecule interaction can promote T cell anergy or deletion by apoptosis [14]. The anergy is characterized by the inability of lymphocytes to produce IL-2, even after an antigenic stimulus, preventing the proliferation of specific clones and the generation of effector cells. Once activated, T cells initiate the proliferation phase, induced by autocrine production of IL-2. At this stage there is an increased expression of the α chain of the IL-2 receptor and increased receptor affinity for the cytokine. After a period of rapid growth, which can range from 3 to 7 days, T lymphocytes differentiate into effector cells that will perform specialized functions, such as helper or cytotoxic T cells [3, 4]. Effector T lymphocytes exhibit a series of phenotypic and functional changes that distinguish them from naïve T lymphocytes. The change in expression of some adhesion molecules, such as decreased CD62L and increased CD2, LFA-1, and CD44, stops the recirculation through the lymph nodes and favors the effector T cell migration to the inflammatory focus and other non-lymphoid tissues.
Cytokines secreted by cells involved in the innate response are crucial in determining a specific “T helper” (Th) response despite many factors, such as the antigen’s route of entry into the body, antigen dose, and co-stimulators, and also influence the adaptive immune response. The CD4+ T cells can differentiate into different subtypes (Th1, Th2, Th3, Th17, regulatory T cells, etc.) according to the pattern of cytokine secretion. Th1 lymphocytes secrete mainly IFN-γ, IL-2, and TNF-α, whereas Th2 lymphocytes produce IL-4, IL-5, and IL-13 [15].
In general, Th1 lymphocytes stimulate a cellular immune response characterized primarily by the activation of macrophages. This process is mediated by soluble or cell surface molecules of T lymphocytes, such as IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF-α, CD40L, and FasL [16]. Activated macrophages produce high levels of oxygen radicals and nitric oxide (NO), antibacterial peptides, and proteases. The activation of these cells increases the expression of MHC-II molecules and B7, CD40, and TNFR receptors, enhancing antigen presentation to T lymphocytes and amplifying the immune response. Th2 lymphocytes produce cytokines that stimulate mainly the humoral immune response, i.e., the production of antibodies. IL-4 induces the switch to IgE, increases the expression of MHC-II molecules on B lymphocytes, stimulates T lymphocytes, increases the proliferation of mast cells, and inhibits macrophage activation. IL-5 stimulates the growth and differentiation of eosinophils [17].
Immune Dysfunction on Perioperative Period
In 1999, the Center for Disease Control and Prevention (CDC) in the USA presented the guideline for prevention of surgical site infection. Surgical site infections are the third most frequently reported nosocomial infection. When surgical patients with nosocomial surgical site infection died, 77 % of the deaths were reported to be related to the infection. Therefore, it is necessary be aware of the major alterations caused in the body by surgery, detectable during the perioperative days. In general, it starts by the vanishing of the skin barrier, which protects against bacterial invasion, and the inflammatory process, which is established during surgery. These alterations affect the innate immunity, the first line of host defense, as much as the adaptive response, during major general surgery [18].
In fact, it has been possible to recognize that even an elective aortic surgery utilizing synthetic graft material, as well as experiments with vascular graft materials in vitro, induce systemic reactions and inflammatory responses [19–25]. Rudensky et al. [26] have attempted to compare the systemic response following two specific cardiac surgery procedures, coronary artery bypass grafting (CABG), versus isolated valve surgery. This study concluded that valve surgery seemed to induce a stronger proinflammatory state than CABG. A significant postoperative increase was observed in both white blood cells (WBC) and monocyte counts, probably reflecting the postoperative inflammatory response invoked by the trauma.
WBCs are the key effector cells that are rapidly mobilized to heal devitalized tissue to prevent secondary microbial invasion (Fig. 3.1a–d). This mobilization is associated with the inflammatory status achieved in order to protect the organism. At this point, numerous alterations due to the damage environment are established. Analysis of the inflammatory response in endovascular stent treatment of aortic aneurysms, for example, also showed elevation of the leukocyte count, which occurred in the premature phase of follow-up, while the lymphocyte and platelet count occurred in a late phase of follow-up. The sedimentation velocity values began to increase 6 h after the surgical procedure, increasing to its peak value in 7 days. The levels of C reactive protein (CRP) increased 6 h after surgery, resulting in a peak value at 48 h. Between 48 h and 7 days, a decrease in serum values was observed, and around the second postoperative month, the CRP values were practically undetectable [27].
The inflammatory state can also be related to the kind and the complexity of the surgery. Major surgery is associated with neutrophil dysfunction, as indicated by the reduced chemotactic ability, phagocytic ability, and superoxide anion production [28, 29]. In addition, surgical injury can lead to a multifactorial perioperative response including activation of complement, coagulation, fibrinolytic, and kallikrein cascades, activation of WBCs with degranulation and protease enzyme release, production of other free radicals, synthesis of various cytokines, and their consequences [30].
In fact, a correlation between the increased level of TNF-α with subsequent significant alteration of cell surface receptors (CD11a, CD11b, CD11c, CD18 and L-selectin) at 60 min post-balloon inflation has been reported. It is necessary to remember that these molecules play an important role in diapedesis, associated with the inflammatory process established after the surgery [31].
Similarly, Schumacher et al. [32] reported an upregulation of adhesion molecules, such as ICAM-1, E-selectin, and P-selectin, and release of cytokines within 12–24 h after implantation of the endovascular device even though a short period of moderate increase of IL-6 and IL-8 has been observed. Asimakopoulis et al. [33] also observed increased CD11b (Mac-1) expression on neutrophils at 24 h post-operation extending to the third postoperative day. CD11b is the intercellular adhesion molecule-1 ligand and could reflect the modulation of this adhesion molecule during the perioperative period. In contrast, Gabriel et al. [27] identified the peak level of ICAM-1 and L-selectin in the first postoperative month and after this period of time a decrease in the levels of both markers. Others demonstrated a significant increase in neutrophil CD11b expression in samples taken 15 min after commencement of surgery, but not in subsequent samples, possibly attributable to marginalization of the activated leucocytes into tissues. In addition, these results demonstrated that the expected inflammatory response is present in both premature and late phases after cardiac surgery, such as implantation of a vascular prosthesis [27]. Together, these results suggest that during the entire inflammatory period the expression of those adhesion molecules will be raised, reflecting increased mobilization of cells between the vessels and the extracellular environment, an action necessary in all phases of the injury caused by the surgery.
Cytokines play an important role in both up- and downregulation of inflammation, and it is involved from the beginning of the innate immune response, trying to stop all injuries and avoiding any infections [34]. In particular, cytokines have been studied as a means to demonstrate the severity of surgical tissue injury.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


