Fig. 7.1
Top panel. Left: pre-procedural angiogram of the right coronary artery showing a 60–70 % mid-vessel stenosis; middle: Intravascular ultrasound (IVUS) examination (4 slices taken at 1-mm distance in the middle of the plaque, with red lines depicting the borders of lumen area, blue lines the external elastic membrane area, and with the difference being the plaque area) shows a large plaque burden with hypo-echogenic features, suspected for a large necrotic core; right: late gadolinium enhancement CMR shows no evidence of pre-existing myocardial necrosis. Bottom panel. Left: angiogram after stenting shows a good result from the PCI; middle: IVUS examination confirms the good post-procedural result, showing a significant reduction of plaque burden; right: Late gadolinium enhancement CMR, however, shows a focal area of endocardial hyper-enhancement in the infero-lateral segment (arrow), suggesting a small myocardial necrosis caused by distal embolization of plaque material during the stenting procedure. The calculated mass of the new necrotic tissue was 2 g. Adapted from Porto I et al. [1]
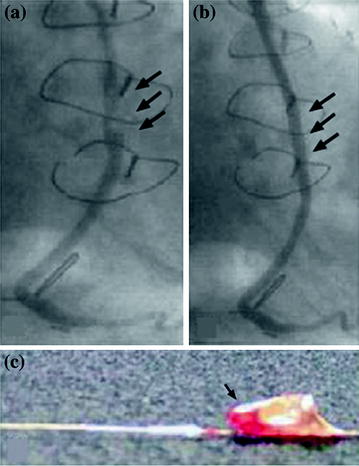
Fig. 7.2
Panel A: coronary angiogram obtained in a 76-year-old patient with an acute coronary syndrome treated for an ulcerated lesion in a saphenous vein graft to the lower circumflex–marginal branch (arrows). A stent was placed in the lesion, with an excellent final result and good distal flow (Panel B, arrows), after distal positioning of a filter basket similar to that shown in Panel C (arrow). Adapted from Kornowski R et al. [4]
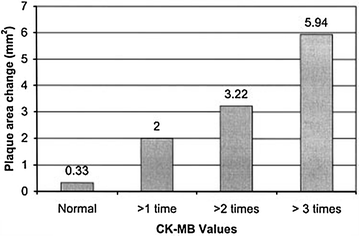
Fig. 7.3
Relation between CK-MB increase after PCI and change in plaque area after stenting assessed by intravascular ultrasound examination. These findings suggest that a considerable amount of the lumen enlargement obtained after stenting is attributable to plaque reduction associated with distal embolization. Adapted from Prati F et al. [5]
Based on these and other observations, the last version of the universal definition of myocardial infarction suggests that, by arbitrary convention, myocardial infarction related to PCI (type 4) should be diagnosed when troponin values >5 times the upper normal limit during the first 48 h after the procedure are detected, occurring from a normal baseline troponin value, together with (1) evidence of prolonged ischemia (≥20 min) as demonstrated by prolonged chest pain, or (2) ischemic ST changes or new pathological Q waves, or (3) angiographic evidence of a flow-limiting complication, such as loss of patency of a side branch, persistent slow-flow or no-reflow, embolization, or (4) imaging evidence of new loss of viable myocardium or new regional wall motion abnormality [6].
The clinical relevance of distal embolization during PCI is considerable. In a recent comprehensive meta-analysis, Testa et al. have assessed the occurrence and the prognostic impact of Tn elevation in patients with normal baseline levels undergoing PCI. Tn elevation after PCI occurred in about one-third of patients. At 18 months follow-up periprocedural Tn elevation was associated with a 50 % increased risk of subsequent major cardiovascular events, a twofold increased risk of death, a threefold increased risk of myocardial infarction, and a 50 % increased risk of re-PCI (Fig. 7.4) [7].
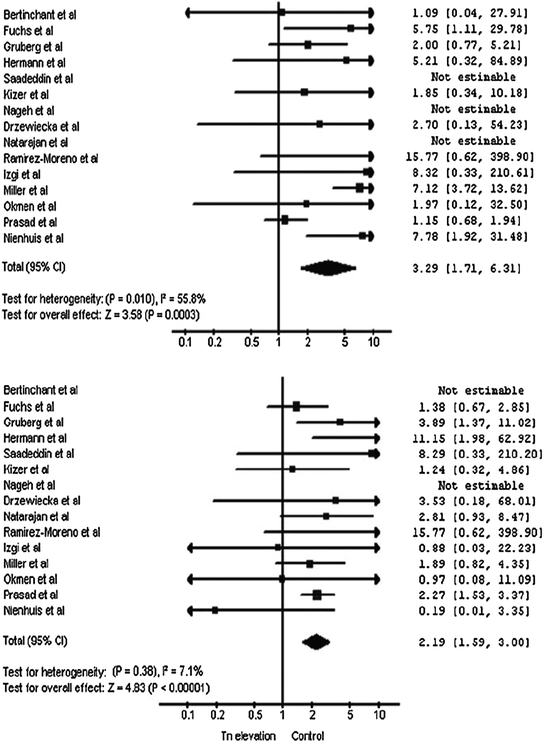
Fig. 7.4
Meta-analysis of studies assessing the impact of troponin raise after PCI on clinical outcome. Top panel. Individual and summary odds ratios (95 % confidence limits) for re-infarction. Bottom panel. Individual and summary odds ratio (95 % confidence limits) for all-cause death. Any troponin raise was associated with a threefold increase of the risk of re-infarction and a twofold increase of the risk of all-cause death. Adapted from Testa L et al. [7]
Although this meta-analysis could not assess the relation between amount of enzyme raise and outcome, this issue has been carefully assessed in a multicenter prospective cohort study, which evaluated the influence of post-procedural elevations of CK-MB on long-term mortality. CK-MB elevation was detected in 16 % of patients (Fig. 7.5) [8]. At 2 years follow-up periprocedural CK-MB elevation was associated with a twofold increased risk of death. A multivariable logistic regression analysis showed that CK-MB elevation independently predicted the risk of death with a linear relationship with the adjusted probability of death. Importantly, when the patients with baseline CK-MB levels above the upper reference limit or procedural complications were removed from the analysis, the CK-MB raise retained a similar independent predictive value. These findings are important because they show that even in patients with apparently successful PCI, a subclinical increase of markers of myocardial necrosis is associated with a higher risk of death at medium long-term follow-up.
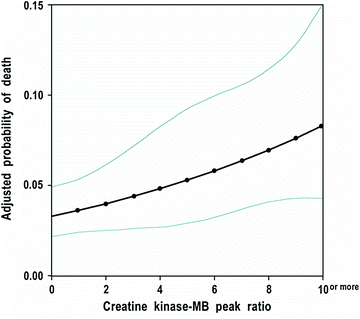
Fig. 7.5
CK-MB peak ratio after PCI and adjusted probability of death 2 years after the procedure. Adapted from Cavallini C et al. [8]
It is only possible to speculate as to why procedural myocardial necrosis marker elevations influence long-term mortality. One potential mechanism may be related to the myocardial damage per se, which might lead to increased mortality as a result of electrical instability or of persistent myocardial ischemia associated with CMD. Alternatively, raised necrosis marker levels after PCI may signal a more active atherosclerotic process associated with an adverse prognosis due to a higher risk of cardiovascular events. It is interesting to note that antithrombotic drugs such as platelet glycoprotein IIb/IIIa receptor inhibitors, which have consistently been shown to reduce post-procedural myocardial damage, also reduce long-term mortality especially in high-risk subsets of patients [9, 10]. This observation supports the notion of a direct effect of periprocedural myocardial necrosis on mortality. It might be speculated that CMD might favor thrombogenic mechanisms in the proximal epicardial coronary arteries (see Chap. 6).
It is worth noting that CMD associated with PCI is characterized not only by distal embolization but also by functional alterations. Indeed, Gregorini et al. found that after stenting CFR transiently decreases in the myocardium supplied by the stented coronary artery branch. She also found that α2-adrenergic blockade by yohimbine normalized CFR [11]. Several mechanisms can be hypothesized to play a role in the periprocedural CFR decrease observed during coronary stenting. In particular, ischemia induced by balloon inflations and the stretch of the artery may elicit a reflex [12] sympathetic increase of α-adrenergic constrictor tone, as observed both in animals and in humans [13–15]. Both α1– and α2-adrenergic receptors are present in the human coronary circulation, with α2-adrenergic receptors being predominant in the microcirculation [14–18].
Accordingly, selective blockade of α1 and α2-adrenergic receptors has different effects on conduit and resistance vessels [19–21]. Notably, Gregorini et al. found that α2-adrenergic receptor blockade, rather than α1 blockade normalized CBF after PCI, thus supporting the notion that this alteration was mediated by coronary microvascular constriction [11].
Taken together, the information on the coronary microvascular effects of PCI shows that while, on the one hand, improvement of myocardial perfusion related to removal of epicardial stenoses is beneficial, on the other hand, the functional and, more importantly, structural microvascular alterations may substantially limit these advantages. Accordingly, in patients with stable angina associated to small or moderate amount of myocardium at risk of myocardial ischemia, stenosis removal by PCI does not improve the outcome. In a recent meta-analysis initial stent implantation for stable CAD showed no evidence of benefit compared with initial medical therapy for prevention of death, nonfatal MI, unplanned revascularization, or angina [22] (see Chap. 6).
The failure of stent implantation to reduce the risk of death or infarction compared with medical therapy in stable CAD reinforces current concepts of the underlying pathophysiologic characteristics of atherosclerosis as a diffuse arterial inflammatory disease that gives rise to vulnerable plaques, the disruption of which leads to coronary thrombosis, myocardial infarction, and death. Lesions most prone to rupture tend to be those of the least hemodynamic consequence, whereas the obstructive lesions that are stented to treat angina or ischemia are paradoxically less prone to rupture [23]. These findings fail to support theories suggesting that PCI might reduce mortality by stabilizing vulnerable plaques [24, 25].
Notably, over 400,000 PCI procedures are performed for the treatment of stable CAD in the United States each year [26]. Despite publication of clinical trials and guidelines supporting the initial use of optimal medical therapy prior to PCI, 44 % of stable patients only receive optimal medical therapy before deciding to perform PCI [27], while at the other extreme approximately 50 % of patients with STEMI do not receive a primary PCI [28]. This resistance to adhere to recommendations derived from high quality evidence is multifactorial. It has been suggested that financial rewards for physicians and hospitals to perform PCI in the fee-for-service health care environment of the United States may contribute to the persistent use of PCI in settings where it has been shown to offer no clinical benefit. In support of this concept, rates of PCI for stable coronary artery disease in Ontario, Canada, where a single-payer government regulates system controls, the annual volume of cardiac procedures, are less than half what they are in the New York State [29]. In the context of controlling rising health care costs, several studies suggest that up to 76 % of patients with stable CAD can avoid PCI altogether if treated with optimal medical therapy [30]. At the other extreme, PCI clearly improves the outcome in patients with ACS. In this setting the benefit of limiting the impending risk of loss of a large amount of myocardium is clearly superior to the detrimental effects of PCI on coronary microcirculation.
7.2 Surgical Interventions
Surgical trauma and cardiopulmonary bypass contribute to a systemic inflammatory response measurable by circulating cytokines [31–35]. This can be the result of many factors, including contact of blood with the bypass circuit, myocardial ischemia during bypass, aortic cross clamping, and reperfusion injury [36–39]. Perioperative myocardial infarction results in further elevation of inflammatory markers [40, 41]. Substantial biomarker elevations after CABG have been shown to have significant prognostic implications [42–45] and usually represent CABG-related myocardial infarction [46–48]. Identifying CABG-related infarction using conventional ECG and biochemical methods, however, can be difficult. Troponins are particularly sensitive biomarkers introduced predominantly for risk stratification in patients with acute coronary syndrome and are the gold standard for identifying myocardial necrosis. However, there is uncertainty about what absolute level of biomarker elevation reflects myocardial infarction after CABG. Furthermore, there are little data on whether an early measurement of biomarkers (at 1 h or less after surgery) can provide any diagnostic or prognostic relevant element. One study using late gadolinium enhancement at CMR imaging and TnI measurement found that about one-third of patients undergoing CABG had new myocardial infarction and that a cut off of TnI of 5 μg/L (upper normal limit 0.6 μg/L) at 1 h had 67 % sensitivity and 79 % specificity for detecting the new infarction. In this study the predictive value of CK-MB for the diagnosis of myocardial infarction was lower than that of TnI: using a cut off of CK-MB of 25 μg/L (upper normal limit 4.8 μg/L) at 1 h had, indeed, 44 % sensitivity and 89 % specificity for detecting the new infarction [49] (Fig. 7.6). Based on these and other observations the last version of the universal definition of myocardial infarction states that by arbitrary convention, myocardial infarction with bypass surgery (type 5) should be diagnosed for troponin values >10 times the upper normal limit during the first 48 h following surgery, occurring from a normal baseline troponin value. In addition, either (1) new pathological Q waves or new left bundle branch block, or (2) angiographically documented new graft or new native coronary artery occlusion, or (3) imaging evidence of new loss of viable myocardium or new regional wall motion abnormality, should be documented for the diagnosis of a CABG-related myocardial infarction [6].
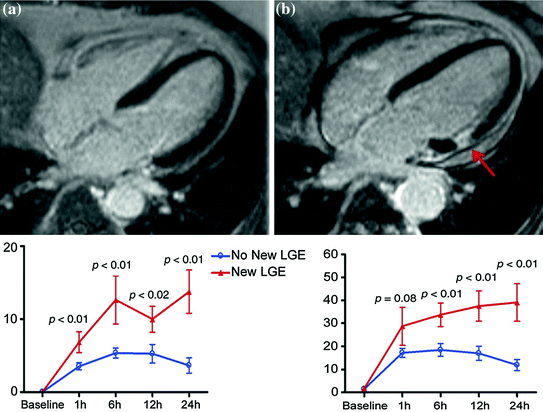
Fig. 7.6
Top panel. a CMR before CABG. b New hyper-enhancement is shown on CMR after CABG (arrow). Bottom panel. Comparison of curves of troponin I release (left) and CK-MB release (right) after CABG in patients with the evidence of new areas of late gadolinium enhancement (LGE, triangles) vs. patients without new LGE areas (no new LGE, circles) at CMR. The error bars are mean standard errors. Adapted from Lim CC et al. [49]
Thus, elevations of biochemical markers of myocardial damage, CK-MB and troponins are common after CABG and have been associated with increased in-hospital and short-term mortality [43, 44, 50, 51]. However, few studies have assessed long-term mortality, especially in relation to troponin peak. Sorras et al., assessed the impact of both CK-MB and TnT on mortality after bypass grafting over 6 years follow-up. They enrolled 1,350 consecutive patients undergoing isolated on-pump coronary bypass grafting and measured CK-MB and TnT at 7, 20 and 44 h postoperatively. Both peak CK-MB and peak TnT independently predicted long-term mortality when analyzed in separate multivariate Cox models, adjusting for baseline demographic characteristics and perioperative risk factors. However, when analyzed simultaneously in the same Cox model, TnT only was a significant predictor. They concluded that both CK-MB and TnT were predictors of mortality after CABG surgery, but TnT appeared to be a better predictor of long-term mortality [52].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


