3.1
Chronic obstructive pulmonary disease
3.2
Interstitial lung disease
3.3
Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4
Sleep-disordered breathing
3.5
Alveolar hypoventilation disorders
3.6
Chronic exposure to high altitude
3.7
Developmental abnormalities
Epidemiology
PH owing to chronic lung disease is the second most common cause of PH in the Western world [5]. Exact determinations of the prevalence of PH in chronic lung disease are difficult due to differences in methodology and definitions employed across the various studies. Recent studies suggest that PH is present in up to 50 % of hospitalized patients with chronic obstructive pulmonary disease (COPD), but in as many as 70–90 % of patients with severe emphysema evaluated for lung volume reduction surgery or lung transplant [6–8]. Similarly, estimates among patients with idiopathic pulmonary fibrosis (IPF) range from 10 to 84 %, with at least 45 % of patients listed for lung transplant being affected [4, 9–11]. The incidence of PH appears even higher at initial diagnosis among patients with combined pulmonary fibrosis and emphysema (CPFE) at 47 %, increasing to 55 % of patients during follow-up [12]. Among patients with obstructive sleep apnea (OSA), 27–42 % of patients have a mean PAP > 20 mmHg, with an even higher prevalence noted in obesity hypoventilation syndrome [13, 14]. High-altitude pulmonary edema (HAPE), a condition characterized by exaggerated hypoxic vasoconstriction and acute PH, is seen in 0.01–0.1 % of visitors of ski resorts in the Rocky Mountains, and in 0.2 % of a general alpine mountaineering population [15, 16]. However, its prevalence can reach up to 7 % in regular mountaineers reaching high altitudes (>4,500 m) within a short period of time, and up to 62 % in predisposed mountaineers [17]. The incidence of PH in patients living at high altitude is not well known. In a Kyrgyz population living at >3,000 m, 20 % of dyspneic patients had hemodynamically confirmed PH [18]. Other studies have shown a prevalence of high-altitude PH between 5 and 18 % in a population living at >3,200 m in South America [19].
Although the mean PAP in group 3 PH is typically between 20 and 35 mmHg, a minority of patients present with a mean PAP greater than 35–40 mmHg, some with relatively preserved lung function [6]. Some experts have labeled this entity PH “out of proportion” to lung disease. For example, this group of patients comprises about 5 % of patients with COPD, and some studies have estimated the prevalence of out of proportion PH in COPD to be similar to the prevalence of idiopathic PAH [6, 20]. Although it is not clear if patients with lung disease and out of proportion PH represent a more permissive phenotype characterized by genetic polymorphisms such as those seen in the serotonin transporter or IL-6 genes [21, 22], or if they represent true group 1 PAH that is superimposed on chronic lung disease, at least epidemiologically true idiopathic PAH may coexist with chronic lung disease.
Pathogenesis
Group 3 PH is pathologically distinct from PAH. The hallmark of PAH is a severe progressive pulmonary hypertensive arteriopathy characterized by plexiform lesions, in situ thrombosis, and extensive arterial wall remodeling and fibrosis [4, 23]. In contrast, the vascular remodeling in group 3 PH is characterized by media hypertrophy, muscularization of small normally nonmuscular arteries, and fibrosis and stiffening of large proximal pulmonary arteries, with notable absence of plexiform lesions (see Fig. 4.1 and Table 4.2). There is also significant vascular inflammation that intensifies the perivascular remodeling [4, 24]. These changes can occur in the presence or absence of hypoxia; in the latter case, factors such as cigarette smoke or mechanical alterations may be the inciting factors (see Fig. 4.2). Technically speaking, the term “hypoxic pulmonary hypertension” is therefore a misnomer; however, since most lung diseases are characterized at least in part by hypoxemia, the term is commonly used.
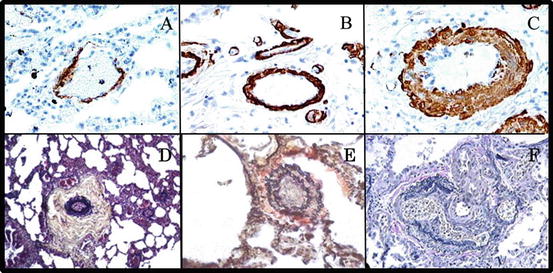
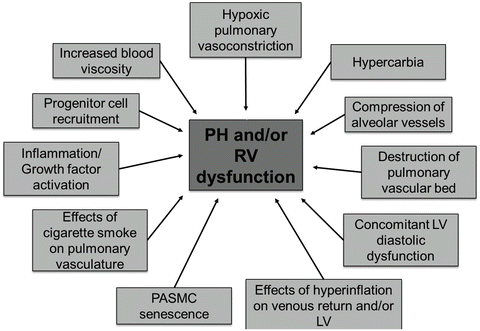
Fig. 4.1
Distinct pulmonary vascular remodeling in group 3 and group 1 pulmonary hypertension. (a) Normal small pulmonary artery. Note thin wall and open lumen. (b, c) Pulmonary arteries from patients with COPD-PH (group 3 PH). Note thickened vascular walls with significant media hypertrophy, especially in (c). However, in both patients, vessel lumens are widely patent. (d–f) Pulmonary vascular remodeling in patients with PAH (group 1 PH). Note significant remodeling of the intima, media, and adventitia (d), with vessel occlusion (e) and plexiform lesions (f). Such massive remodeling is not observed in patients with group 3 PH. (a–c) Reproduced with permission from [21]; D-F reproduced with permission from [4]
Muscularization of previously nonmuscularized arterioles |
Medial hypertrophy of muscularized arteries (particularly in smaller branches) |
Longitudinally oriented intimal smooth muscle cells |
Mild medial hypertrophy of veins |
Perivascular inflammatory infiltrates |
In patients with interstitial lung disease: Eccentric intimal fibrosis of arteries (and to a lesser degree of veins) |
Fig. 4.2
Mechanisms of PH and RV dysfunction in HPH. Note multifactorial mechanism of PH development and/or RV dysfunction (cor pulmonale). Contribution of the listed factors may vary depending on type and severity of the underlying lung disease, disease stage (early vs. late), comorbidities, ongoing exposures, and genetic predisposition. LV left ventricle, PASMC pulmonary artery smooth muscle cells, PH pulmonary hypertension, RV right ventricle
The Effects of Hypoxia on the Pulmonary Vasculature
Hypoxia has an immediate effect on PAP via hypoxic pulmonary vasoconstriction (HPV). In the setting of sustained exposure, there is also a delayed effect via hypoxia-induced pulmonary vascular remodeling.
Hypoxic Pulmonary Vasoconstriction (HPV)
HPV refers to a process in which the pulmonary vasculature responds to a hypoxic stimulus with a vasoconstrictor response. This process, which is unique to the pulmonary vasculature (systemic vessels dilate upon hypoxia exposure), is thought to be a protective reflex in order to maintain ventilation and perfusion matching in the setting of focal lung disease such as consolidation [25, 26]. However, if the entire pulmonary vasculature constricts due to global hypoxia exposure, a significant increase in pulmonary vascular resistance (PVR) and thus right ventricular (RV) afterload ensues.
Excessive HPV is also the culprit of high-altitude pulmonary edema (HAPE), a potentially lethal complication in susceptible individuals visiting altitudes >2,500 m [17]. HAPE is characterized by exaggerated vasoconstriction, increased PA pressures, excessive shear stress, and subsequent stress fracture of the pulmonary vascular endothelium [27–30]. The latter appears to occur as the result of perfusion heterogeneity within the lung caused by uneven distribution of the severity of the HPV response [31]. This results in areas of the lung in which blood flow is severely diminished and redirected to areas where HPV is less intense. These high flow areas are prone to capillary failure leading to the patchy distribution of pulmonary edema formation that is characteristic of HAPE (Fig. 4.3) [32]. Rapid ascent and exercise at high altitude are risk factors, as are conditions causing a restricted pulmonary vascular bed (e.g., unilateral absence of a pulmonary artery) [33, 34].
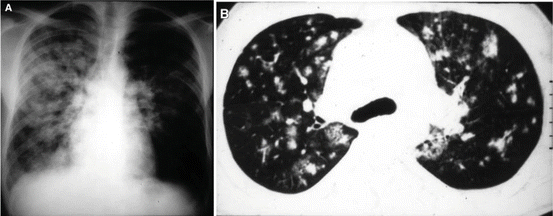
Fig. 4.3
High-altitude pulmonary edema. (a) Predominant right-sided patchy airspace disease in a 37-year-old mountaineer with high-altitude pulmonary edema (HAPE). (b) Chest CT of a 27-year-old mountaineer with history of recurrent HAPE demonstrating nondependent, patchy airspace disease. Note areas of normal lung adjacent to areas of dense infiltrate (with permission from reference 30b)
Even though first described many years ago, the exact mechanisms of HPV are still incompletely understood [33]. Recent research implicates mitochondria in pulmonary artery smooth muscle cells as sensors of hypoxia and effectors of HPV, with hypoxia-induced changes in mitochondrial concentration of reactive oxygen species (ROS) leading to inhibition of membrane potassium channels (e.g., Kv1.5, Kv2.1), depolarization of membrane potential, opening of voltage-dependent calcium channels, increases in intracellular calcium concentration, and subsequent vasoconstriction [25, 26, 35–39].
Hypoxic Pulmonary Vascular Remodeling
Chronic hypoxia induces significant structural remodeling characterized by the hallmark appearance of smooth muscle-like cells in previously nonmuscularized pulmonary vessels. This is demonstrated even in otherwise healthy persons chronically living at altitude, who have an increased number of muscularized peripheral pulmonary arterial branches associated with increased PAP at rest and with exertion [40, 41]. In humans, after as little as 6 weeks of chronic hypoxia exposure, changes in PVR are not immediately reversible with administration of oxygen, suggesting significant remodeling has already taken place [42]. Importantly, and in striking contrast to PAH, the hypoxia-induced pulmonary artery remodeling is partially to fully reversible upon cessation of the hypoxic stimulus (e.g., moving to a lower altitude) [41, 43].
All portions of the pulmonary arterial wall are involved in hypoxic pulmonary vascular remodeling. Major contributors to the development of HPH are the hypoxia-inducible factors (HIFs), in particular HIF-1α and HIF-2α [44, 45]. HIFs function as transcription factors that bind to specific hypoxia-responsive elements of their target genes (e.g., erythropoietin, vascular endothelial growth factor, Glut-1, endothelin-1, angiopoietin-2), thus regulating almost every single process affected by hypoxia. In the pulmonary vasculature, HIF-1α appears to play a more predominant role in smooth muscle cells, while HIF-2α is primary located in pulmonary artery endothelial cells [44, 45]. The critical role of HIFs in the development of HPH was demonstrated by an elegant animal study, in which mice partially deficient in HIF-1α had an attenuated response to hypoxia and were largely protected from HPH [46]. Interestingly, genetic variations in the HIF system are associated with better adaptation to high altitude. For example, several studies have recently demonstrated that Tibetan highlanders, a population that has previously been shown to exhibit better adaptation to high altitude than other populations living at similar altitude, exhibit single nucleotide polymorphisms (SNPs) in the gene encoding for HIF-2α, as well as in genes encoding for regulators of HIF-1α signaling [47–49].
On a cellular level, hypoxic pulmonary vascular remodeling is characterized by activation and involvement of all cell types of the pulmonary vasculature. In particular, the hypoxic pulmonary vasculature is characterized by endothelial cell activation, smooth muscle cell proliferation, de-differentiation of adventitial fibroblasts into myofibroblasts, activation and recruitment of inflammatory cells and progenitor cells, and increased collagen production [3, 4, 50].
Fibroblasts within the adventitia are among the first cells to be activated by hypoxia and vascular stress, resulting in increased expression of proinflammatory cytokines (e.g., IL-1β, IL-6, CCL2, CXCL12, VCAM-1) and cellular proliferation and differentiation into myofibroblasts, with increased matrix protein production and deposition of collagen and elastin, as well as migration of cells into the media and even intima [3, 24]. An influx of macrophages, fibroblasts, and myofibroblasts, as well as resident and circulating progenitor cells, contributes to the intimal hyperplasia found in HPH. Medial hypertrophy and muscularization of previously nonmuscularized arterioles are driven by the same processes, as well as by smooth muscle cell hypertrophy and proliferation.
Endothelial cells, while not significantly proliferating after exposure to low oxygen levels, contribute to hypoxic pulmonary vascular remodeling through cell surface activation and secretion of paracrine factors, thus resulting in smooth muscle cell proliferation, as well as recruitment of progenitor and proinflammatory cells. In particular, hypoxic endothelial cell activation results in increased production of vasoconstrictors and growth factors (PDGF-β, IGF, VEGF, bFGF, serotonin), adhesion molecules (P-selectin, ICAM, VCAM), cytokines (IL-1, IL-8), procoagulants (tissue factor, PAI-1), and matrix molecules (laminin, fibronectin) [3, 21, 50].
In addition to external and paracrine factors, smooth muscle cells within the media are also stimulated to proliferate by various intracellular signaling mechanisms. For example, changes in mitochondrial redox potential lead to inhibition of potassium channels, activation of voltage-dependent calcium channels, and calcium influx, resulting in both vasoconstriction and smooth muscle cell proliferation [25, 26, 35–39, 51]. Together, the cells of the pulmonary artery wall serve to recruit and activate circulating inflammatory cells such as monocytes and fibrocytes (predominantly via the vasa vasorum), as well as resident and circulating progenitor cells, thus further contributing to ongoing chronic inflammation, vasoconstriction, and structural remodeling [3, 24, 52]. In this context, it is important to note that upon chronic hypoxia exposure, alveolar inflammation seems to precede pulmonary vascular inflammation, and amelioration of the inflammatory process in the alveolar space is associated with decreased pulmonary vascular remodeling and HPH [53, 54].
Hypoxia-Independent Mechanisms of Pulmonary Vascular Remodeling
Pulmonary vasoconstriction and activation of the pulmonary vascular remodeling process may also occur independently of hypoxia. For example, hypoxia-independent factors implicated in PH pathogenesis in chronic lung disease include vasoconstrictive effects of hypercarbia [55], compression and destruction of alveolar vessels from structural alterations and fibrotic lung disease [56–58], concomitant left ventricular systolic or diastolic dysfunction [59, 60], hemodynamic effects of hyperinflation on pulmonary vascular filling and RV or left ventricular function [61, 62], as well as pulmonary artery smooth muscle cell senescence [63, 64] and toxic effects of cigarette smoke (see Fig. 4.2) [65–68].
Pulmonary artery wall cell senescence has recently been identified as a potential contributor to PH in COPD [63, 64]. This paradigm encompasses a scenario where senescent pulmonary artery smooth muscle cells from COPD patients (characterized by increased p16, p21 and β-galactosidase expression, fewer cell population doublings, and shorter telomeres than cells from controls) stimulate growth and migration of adjacent normal smooth muscle cells through the production and release of paracrine factors such as IL-6, IL-8, TNF-α, MCP-1, and TGF-β [63]. This notion is supported by the fact that the senescent cells are almost exclusively confined to the media, thus being adjacent to areas of marked cell proliferation [63]. Interestingly, this paradigm of telomere shortening, premature senescence, and proinflammatory signaling has also been described for pulmonary artery endothelial cells in COPD patients [64]. However, even though there is an association between senescence and pulmonary vascular remodeling, as well as an inverse relationship between telomere length and mean PAP and PVR [63], it currently remains unknown if senescence is a cause or a consequence of the pulmonary vascular remodeling observed in COPD. In addition, the potential triggers for pulmonary artery smooth muscle cell and endothelial cell senescence (e.g., age, hypoxia, inflammation, oxidative stress) have not yet been identified [69].
Importantly, recent studies implicated chronic exposure to cigarette smoke as a major contributor to PH development. In particular, cigarette smoke has been shown to result in endothelial cell dysfunction with a subsequent vasodilator-vasoconstrictor imbalance due to decreased nitric oxide and prostacyclin and increased ET-1. This is associated with smooth muscle cell proliferation, progenitor and inflammatory cell recruitment, and distortion of the normal pulmonary wall architecture [65–68]. Oxidative stress, reactive nitrogen species, and inflammation have been implicated as major mediators of cigarette smoke-induced vascular damage [65, 68, 70–72]. Such changes may be seen even in smokers without overt emphysema. For example, an intriguing recent study demonstrated that in the setting of chronic cigarette smoke exposure, pulmonary vascular dysfunction and PH can precede alveolar destruction and emphysema [70]. The authors showed that cigarette smoke causes endothelial dysfunction with increased inducible nitric oxide synthase (iNOS), inflammation, and smooth muscle cell proliferation, clinically resulting in RV hypertrophy and PH [67]. Interestingly, development of PH in this model was dependent on iNOS from bone marrow-derived cells [70].
Right Ventricular Dysfunction in Chronic Lung Disease (Cor Pulmonale)
The vascular remodeling and inflammation in HPH ultimately lead to pressure overload of the RV, complicated by RV hypertrophy, remodeling, and ultimately death. Elevated RV afterload is reflected by a steady increase in the resistance of the pulmonary vascular bed, by increased blood viscosity from elevated red blood cell mass, and by decreased dynamic compliance and stiffening of the large proximal pulmonary arteries [4, 73]. The initial response of the RV is an adaptive remodeling, characterized by increased capillarization and cardiac myocyte hypertrophy, with absence of apoptosis and fibrosis [74]. RV contractility is relatively preserved, but the RV is prone to dysfunction during episodes of further hypoxemia or air trapping, which may occur during pulmonary exacerbations, exercise, or nocturnal desaturations [75]. While RV dysfunction in these settings most likely is due to increases in afterload, thoracic hyperinflation (as seen in emphysema or bullous lung disease) may further decrease RV stroke volume by decreasing cardiac preload [61].
Importantly in HPH, the degree of RV hypertrophy correlates with the severity of hypoxemia. Hypoxia-induced RV hypertrophy occurs in conjunction with increases in gene expression of mRNA encoding for proinflammatory and chemotactic cytokines (e.g., IL-1β, S100A4, MCP-1, and SDF-1) [76]. The end result is cor pulmonale, characterized by RV hypertrophy, dilatation, and dysfunction in the setting of chronic lung disease and HPH.
Systemic factors such as neurohormonal activation further contribute to RV dysfunction and fluid retention. Neurohormonal activation is particularly pronounced in the setting of hypercarbic states, as can be seen in end-stage COPD, severe restrictive lung disease, and obesity hypoventilation syndrome [77]. In these conditions, the combination of hypercarbia and hypoxia results in decreased effective renal plasma flow, increased activation of the renin-aldosterone system, and increased production of vasopressin [78]. The result is excessive sodium and water retention, leading to edema and further increases in preload and afterload, ultimately further exacerbating RV dysfunction [75, 77].
Clinical Presentation, Relevance, and Prognostic Implications
Pathologic pulmonary vascular abnormalities precede the development of clinically apparent HPH. Initially, elevated PAP may only be seen during exercise, exacerbations, or nocturnal desaturations, and the presence of exercise-induced PH is a strong predictor for later development of resting PH [79]. The clinical presentation of HPH may be difficult to distinguish from the associated pulmonary disease, as dyspnea, fatigue, cough, chest pain, and edema may all be due to the underlying lung disease. Thus, a high index of suspicion is required. One notable difference is the presentation of “out of proportion” PH. Although most patients with group 3 PH have mild-to-moderate increases in PAP with mean PAP typically not exceeding 35–40 mmHg, a small percentage (<5 % of COPD patients) present with mean PAP greater than 40 mmHg [6]. These patients often exhibit only mild-to-moderate airflow obstruction but significant impairments in diffusing capacity, as well as more severe hypoxemia and hypocarbia [6]. The extent of pulmonary arterial lesions in explanted lungs after transplantation correlates with the severity of pulmonary hypertension in COPD, with progressively severe medial hypertrophy and intimal fibrosis [80].
Similar to the COPD-PH population, PH in the setting of pulmonary fibrosis usually falls into the mild-to-moderate range. For example, in a study of IPF patients evaluated for lung transplantation, median mean PAP was 31 mmHg, with an interquartile range of 28–38 mmHg [81]. PH appears to be more pronounced, however, in the syndrome of CPFE. In a series of 40 patients with CPFE and PH, mean PAP was 40 ± 9 mmHg, cardiac index was 2.5 ± 0.7 L/min/m2, and PVR was 521 ± 205 dyn s cm−5 [82].
Finally, among patients with sleep-disordered breathing, mean PAP was found to be 28 ± 6 mmHg, and cardiac output was maintained, again indicating that group 3 is usually mild to moderate [13]. Higher body mass index, higher daytime carbon dioxide tension, and lower daytime oxygen tension are strongly correlated with the development of PH in this setting [13]. Among 26 patients with obesity hypoventilation syndrome undergoing evaluation for bariatric surgery, mean PAP was 36 ± 14 mmHg, compared to 18 ± 6 mmHg in 20 obese patients without hypoventilation. The higher elevations in PAP in this study may be in part due to a high incidence of diastolic dysfunction, which was frequently identified in this study [83].
Regardless of the cause, even mild PH in the setting of chronic lung disease is associated with poorer clinical outcomes. For example, there are significant increases in the frequency of pulmonary exacerbations and hospitalizations in COPD patients with mean PAP above 18 mmHg [84]. Furthermore, patients with COPD or IPF and concomitant PH have poorer exercise capacity [58]. A recent study of COPD patients with mild, moderate, or severe PH further investigated this phenomenon and demonstrated that COPD patients with mild or moderate PH exhibit ventilatory limitations during exercise, while patients with severe PH are characterized by circulatory limitations, as evidenced by decreased cardiac output and central venous oxygen saturation [85]. Lastly, for those patients who require lung transplant, there is an increased risk of primary graft dysfunction, with a 1.6-fold increased risk for every 10 mmHg increase in mean PAP [86].
Importantly, PH and RV dysfunction in the setting of chronic lung disease are associated with worse survival, with increasing mortality correlating with the severity of elevation in mean PAP [11, 56, 73, 82, 87, 88]. In fact, the best prognostic factor in COPD patients requiring long-term home oxygen therapy was not the forced expiratory volume, hypoxemia, or hypercapnia but the degree of PH [89]. Similar findings were observed in a recent study of IPF patients referred for lung transplantation. In this cohort, echocardiographically determined RV size and RV dysfunction, as well as higher PVR, were independent predictors of mortality [88].
Diagnosis
Diagnostic tools for the detection of group 3 PH do not differ substantially from those used in group 1 PH, but there are some special considerations in patients with chronic lung disease. Unfortunately, clinical examination is insensitive in diagnosing group 3 PH. A loud second heart sound or tricuspid regurgitation may be obscured by hyperinflation or adventitious lung sounds, and edema, though indicating the presence of cor pulmonale, is a late finding in HPH.
Routine pulmonary diagnostics such as electrocardiogram, pulmonary function testing, 6 min walk testing, and brain natriuretic peptide level determination may provide important clues for the diagnosis. Although an electrocardiogram showing right atrial and RV hypertrophy and RV strain is fairly specific for PH, the absence of these findings does not preclude a diagnosis of PH. On pulmonary function testing, diffusing capacity is frequently decreased out of proportion to the decrease in the forced expiratory volume or forced vital capacity, particularly in patients with PH out of proportion to their underlying disease or in CPFE [82]. Significant desaturations during 6 min walk testing suggest an inadequate cardiopulmonary reserve and point towards PH [6]. Similarly, profound hypoxemia at rest may be a sign of significant PH. Brain natriuretic peptide levels, when elevated in the absence of left heart disease, renal insufficiency, or pulmonary embolism, may serve as an indicator of RV strain and as a prognostic marker for mortality [90].
Standard CT imaging may show evidence of RV dysfunction, such as an enlarged RV with a right ventricle-to-left ventricle ratio greater than one, a dilated pulmonary artery, or reflux of intravenous contrast into the inferior vena cava and hepatic veins indicative of tricuspid regurgitation (see Fig. 4.4). The presence of an increased pulmonary artery diameter on routine chest CT imaging has recently been identified as a predictor of exacerbations in patients with COPD [91]. However, pulmonary artery enlargement (especially if mild to moderate) is not specific for the presence of group 3 PH and may indicate PAP increases from volume overload, left heart disease, pulmonary embolism, or sleep apnea [92].
Echocardiography is an important screening tool in patients with lung disease and is critical for detecting RV structural abnormalities. Unfortunately, echocardiography is less accurate for the estimation of PAP in patients with chronic lung disease. A cohort study of 374 lung transplant candidates showed a sensitivity and specificity of only 85 % and 55 %, respectively, for diagnosis of PH, with 52 % of RV systolic pressure measurements being inaccurate by >10 mmHg [93]. This inaccuracy is due at least in part to poor echocardiographic windows and inadequate visualization of the tricuspid regurgitant jet due to lung hyperinflation. Consequentially, PH should be suspected if there is echocardiographic evidence of right heart chamber enlargement, leftward septal shift, and/or RV hypokinesis, even if the RV systolic pressure is not significantly elevated or not measurable. The role of newer echocardiographic methods such as tissue Doppler or speckle tracking for assessment of cor pulmonale and group 3 PH has not been assessed in detail, but studies in patients with PAH suggest that these are sensitive methods for the assessment of RV function [94–97]. However, limitations with regard to lung hyperinflation may apply to these techniques as well.
Cardiac MRI, though limited by availability and cost, is increasingly being used for determination of RV form and function [98, 99]. While there is no role for routine MRI scanning of the RV in chronic lung disease at this point, cardiac MRI should be considered if an accurate assessment of RV form and function is required and the RV cannot be visualized adequately on echocardiography.
As with other forms of PH, right heart catheterization (RHC) remains the gold standard for diagnosis of HPH. RHC should be considered in patients with significant PH risk factors, such as otherwise unexplained dyspnea, significant hypoxemia, desaturations during 6 min walk testing, elevated brain natriuretic peptide levels, or isolated decreases in DLCO. Similarly, RHC is indicated if there is echocardiographic evidence of significant PH. Since the presence of PH increases the risk of COPD exacerbations, RHC should also be considered in patients with recurrent admissions for COPD exacerbation and/or cor pulmonale [84]. However, it is important to emphasize that other etiologies of dyspnea and exacerbations commonly encountered in chronic lung disease need to be ruled out before proceeding with RHC, including venous thromboembolism, coronary artery disease, ongoing tobacco abuse, medical nonadherence, or infection with nontuberculous mycobacteria. Similarly, treatment for the underlying lung disease and/or hypoxemia should be optimized as much as possible before RHC is considered. Lastly, RHC in the setting of an acute exacerbation of the underlying lung disease yields little information about the patient’s chronic state, as PA pressures may be temporarily elevated due to hypoxemia, hypercarbia, or volume overload.
In addition to quantifying the severity of PH, RHC may help exclude other causes of PH. Hemodynamic assessment during RHC should reveal a mean PAP ≥ 25 mmHg and a pulmonary capillary wedge pressure ≤15 mmHg to confirm group 3 PH. Borderline or elevated pulmonary capillary wedge pressures may suggest concomitant systolic or diastolic heart disease, and can be further assessed by measuring a concomitant left ventricular end-diastolic pressure or by reassessing hemodynamics after a saline bolus or exercise challenge [100]. Typically, PVR and transpulmonary pressure gradient (mPAP-PCWP) are low (≤3 Wood units and ≤12 mmHg, respectively). However, in the setting of out of proportion PH, or in the presence of other comorbidities known to cause PH (e.g., sleep-disordered breathing, pulmonary emboli, or left heart disease), both parameters may be markedly elevated.
Of note, patients with severe dyspnea or obesity may exhibit significant intrathoracic pressure changes due to increased respiratory efforts [101]. This may lead to artifactual decreases in hemodynamic parameters if software-generated pressure readings are used, as those values simply represent an automated mean of the pressure readings [102]. It is therefore important to emphasize that all pressures should be determined at end-expiration with the patient breathing comfortably [101]. One exception to this paradigm applies to patients with significant dynamic hyperinflation and air trapping, in whom end-expiratory pressures may be falsely elevated, and thus in these patients pressures should be determined as the mean over several respiratory cycles.
Treatment
General Treatment Strategies
Therapeutic strategies for group 3 PH focus on aggressively treating the underlying condition causing the elevated PAP. In hypoxemic patients with severe COPD, continuous long-term oxygen therapy is associated with improvement in survival irrespective of the presence of PH [103]. However, among patients with concomitant PH, oxygen therapy for greater than 18 h per day was shown to decrease resting PAP by 3 mmHg and exercise PAP by 6 mmHg. On the other hand, the same study showed that nocturnal oxygen therapy alone was not sufficient to improve mortality [103]. It is currently recommended that hypoxemia during exercise be corrected with the use of oxygen supplementation, even though the evidence supporting this approach is less robust.
Smoking cessation is critical to attenuating the ongoing endothelial dysfunction and inflammation that promote pulmonary vascular remodeling and PH development from tobacco exposure. Recent studies show that cigarette smoke can induce PH though iNOS activation, even before the parenchymal changes of emphysema develop [70]. Similarly, smoking cessation also prevents further parenchymal damage as a contributor to PH development.
Pulmonary rehabilitation may be beneficial in HPH, though special considerations are required. Symptoms can help determine a safe level of submaximal exercise, and patients should avoid activities that cause symptoms such as dizziness, presyncope, and chest pain. Exercises such as heavy lifting, valsalva maneuvers, or interval training should be avoided due to potential rapid changes in cardiopulmonary hemodynamics [104].
Due to the high incidence of sleep-disordered breathing among patients with HPH, polysomnography should be considered in all patients with sleep-disordered breathing symptoms, including morning headaches, excessive fatigue, or witnessed apneas. Patients with both COPD and OSA have significantly higher mortality and risk of hospitalization than patients with either COPD or OSA alone, and both hospitalizations and mortality are ameliorated by the use of positive airway pressure [105]. In OSA, the use of continuous positive airway pressure begins improving RV end-diastolic diameter and RV systolic pressure in as little as 3 months, with continued cardiac remodeling with long-term use [106]. Patients with obesity hypoventilation syndrome benefit from noninvasive positive pressure ventilation and weight loss, including bariatric surgery [83].
Even though there is a general lack of published and evidence-based strategies, clinical experience suggests that diuretics are indicated if there is clinical, echocardiographic, or hemodynamic evidence of elevated right atrial pressures. Loop diuretics such as furosemide are generally preferred. Although aldosterone antagonists are conceptually appealing due to inhibition of the renin-aldosterone system, there are no studies in HPH to guide therapy. Significant diuresis is frequently required, though caution must be taken to avoid over diuresis [75]. When indicated, the use of continuous positive airway pressure or noninvasive positive pressure ventilation may help with fluid mobilization.
Lastly, given the reversibility of hypoxia-induced pulmonary vascular remodeling upon exposure to higher alveolar oxygen pressures, patients with HPH living at high altitude are recommended to move to lower altitudes [4]. If such an approach is not feasible, an alternative but technically much more challenging strategy encompasses oxygen enrichment of the ambient air [41]. HAPE is treated with descent to lower altitudes, oxygen, and nifedipine [41].
Pulmonary Vasodilators
Given the development of several new drugs in PAH over the past decade, there has been significant excitement to translate these medications into use within group 3 PH. Unfortunately, this excitement has been met largely with disappointment, likely because pulmonary vasodilators may inhibit HPV, resulting in increased ventilation-perfusion mismatch and impaired gas exchange. As such, a clear role for pulmonary vasodilators in group 3 PH has not yet been established, and the general use of PAH-specific therapies in this patient population is currently not recommended. Studies of pulmonary vasodilator use in group 3 PH are reviewed in detail below and in Table 4.3.
Table 4.3
Overview of studies of PAH-specific therapies in WHO group 3 PH
Study | Design | Patient population | Study size | Study duration | Medication | Outcome |
|---|---|---|---|---|---|---|
Stolz, ERJ (2008) | 2:1 Randomized double blind placebo controlled | Severe to very severe COPD and mild PH by ECHO | 30 | 12 weeks | Bosentan 62.5 mg bid orally, increased to 125 mg bid after 2 weeks | No change in exercise capacity, ↓ oxygenation, ↓QOL |
Dernaika, Respiration (2010) | Cohort single treatment
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|