Fig. 1
(a) Impairment of hypoxia-induced endothelium-dependent vasorelaxation compared to control in early preatherosclerosis, *p < 0.05; (b) No significant influence of intermittent hypoxia on endothelium-associated vasorelaxation in advanced preatherosclerosis
Consequently, levels of endothelial microparticles which mirror the degree of endothelial destruction were higher under hypoxia vs. control only at early stages of vascular disease (0.28 ± 0.05 % and 0.15 ± 0.02 %, respectively, p < 0.05, Fig. 2), without showing any relevant differences between these groups in more advanced vasculopathy (0.92 ± 0.61 % and 1.03 ± 0.33 %, respectively, p > 0.05).
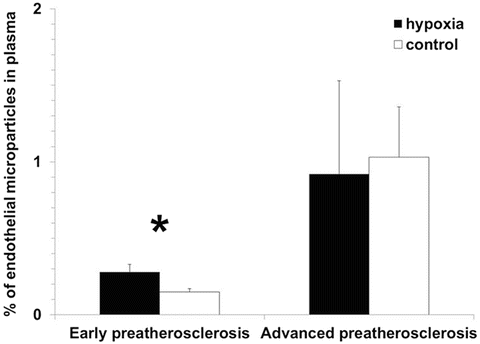
Fig. 2
Higher levels of endothelial microparticles in blood plasma under intermittent hypoxia compared to control in early preatherosclerosis. No relevant differences were noted in the percentage of endothelial microparticles between hypoxia and control group in advanced preatherosclerosis, *p < 0.05
The analysis of compensatory pathomechanisms involving the presence of endothelial progenitor cells in bone marrow and blood showed significant changes in the number of these cells under hypoxia vs. control in early preatherosclerosis. Specifically, the percentage of EPCs increased in bone marrow (2.0 ± 0.4 % and 1.1 ± 0.2 %, respectively, p < 0.05, Fig. 3) and decreased in blood (2.0 ± 0.5 % and 5.3 ± 1.9 %, respectively, p < 0.05, Fig. 4) under hypoxia compared to control. Such alternations were not observed during a further time course of endothelial dysfunction (bone marrow: 1.2 ± 0.3 % and 0.9 ± 0.2 %, respectively, p > 0.05; blood: 1.6 ± 0.3 % and 1.6 ± 0.2 %, respectively, p > 0.05).
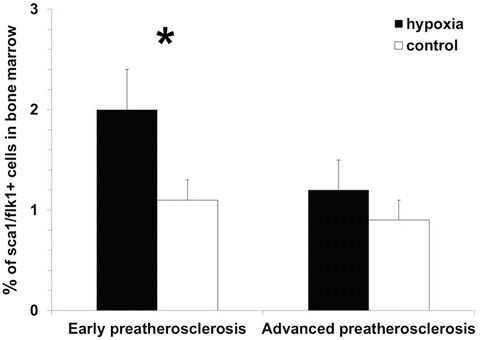
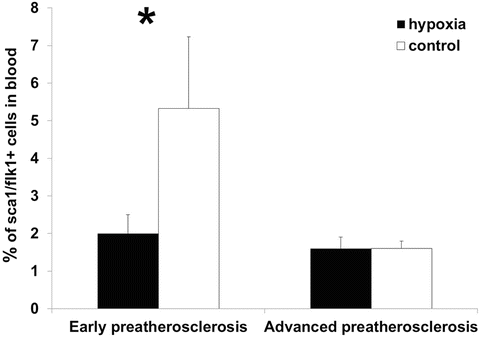
Fig. 3
Elevated numbers of endothelial progenitor cells in bone marrow under intermittent hypoxia vs. control in early preatherosclerosis in contrast to similar levels of these cells in both groups in advanced preatherosclerosis, *p < 0.05
Fig. 4
Reduced levels of endothelial progenitor cells in blood under hypoxia compared to control in early preatherosclerosis. No significant differences in percentage of these cells between both groups in advanced preatherosclerosis, *p < 0.05
4 Discussion
This study demonstrates the influence of intermittent hypoxia on endothelial dysfunction and endothelial repair capacity depending on the degree of advancement of thoracic artery preatherosclerosis. In particular, we showed that intermittent hypoxia decreased endothelial function at early stage of vascular disease. Similar results have been obtained in animal models and human trials (Dematteis et al. 2008; Phillips et al. 2004; Ip et al. 2004). However, dependence of the hypoxia-induced endothelial dysfunction on the initial extent of vasculopathy has not been specifically considered. In the present work, we showed that intermittent hypoxia impairs endothelial function mainly at the very beginning of vascular disease. During a further time course of vascular disorder the negative effects of hypoxia on endothelium are limited. It seems that additional deleterious stimulus such as hypoxia play a rather subordinated role in the worsening of endothelial function once the process of endothelial damage has reached an advanced stage. In line with the above data, we identified higher levels of endothelial microparticles in blood reflecting vascular injury under hypoxia vs. control in initial phases of vasculopathy. As endothelial dysfunction progressed, the overall percentage of microparticles increased, but hypoxia did not significantly augment the levels of endothelial microparticles compared to control. This finding may at least in part explain some previous apparently contradictory data demonstrating enhanced or unchanged endothelial microparticle numbers under hypoxia in comparison to control (Ayers et al. 2009; Akinnusi and El Solh 2009), possibly resulting from investigating of microparticles under hypoxic conditions in different stages of vascular pathology.
The next issue of the present study was the analysis of endothelial repair capacity mediated by endothelial progenitor cells. The number of endothelial progenitor cells increased in bone marrow under hypoxia in early preatherosclerosis. In contrast, the percentage of these cells was lower in blood in the hypoxia vs. control group. These findings point to the hypoxia-induced activation of central compensatory mechanisms. However, the peripheral repair capacity in blood seems to be attenuated under hypoxia. The last result may be explained due to the reduced matrix metalloproteinase-9-dependent release of endothelial progenitor cells from bone marrow to the blood, which has been shown by our group in a previous work (Tuleta et al. 2014). Interestingly, the levels of blood EPCs decreased under normoxia as endothelial function worsened suggesting progressive depletion of repair mechanisms. Hypoxic conditions had no relevant influence on the number of these cells in bone marrow or blood in the stage of advanced endothelial dysfunction. Thus, our data showing different responses of EPCs to the hypoxic stimulus depending on the severity of vascular disease may contribute to the explanation of the fact that several studies concerning circulating endothelial progenitor cell levels provide inconsistent results (Jelic et al. 2008; Kizawa et al. 2009). This is probably due to the investigation of populations with different extent of preatherosclerotic vessel changes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


