Chapter 24 Hypoxia
 Intracellular acidosis from anaerobic metabolism occurs soon after the onset of cellular hypoxia, and is worse when there is a plentiful supply of glucose to the cell.
Intracellular acidosis from anaerobic metabolism occurs soon after the onset of cellular hypoxia, and is worse when there is a plentiful supply of glucose to the cell.
 In nervous tissue the uncontrolled release of excitatory amino acids exacerbates the hypoxic damage.
In nervous tissue the uncontrolled release of excitatory amino acids exacerbates the hypoxic damage.
Chapter 1 explained how all but the simplest forms of life have evolved to exploit the immense advantages of oxidative metabolism. The price they have paid is to become dependent on oxygen for their survival. The essential feature of hypoxia is the cessation of oxidative phosphorylation (page 199) when the mitochondrial Po2 falls below a critical level. Anaerobic pathways, in particular the glycolytic pathway (see Figure 11.13), then come into play. These trigger a complex series of cellular changes leading first to reduced cellular function and ultimately to cell death.
Biochemical Changes in Hypoxia
Depletion of High-Energy Compounds
Anaerobic metabolism produces only one-nineteenth of the yield of the high-energy phosphate compound adenosine triphosphate (ATP) per mole of glucose, when compared with aerobic metabolism (page 200). In organs with a high metabolic rate such as the brain, it is impossible to increase glucose transport sufficiently to maintain the normal level of ATP production. Therefore, during hypoxia, the ATP/ADP ratio falls and there is a rapid decline in the level of all high energy compounds (Figure 24.1). Very similar changes occur in response to arterial hypotension. These changes will rapidly block cerebral function, but organs with a lower energy requirement will continue to function for a longer time and are thus more resistant to hypoxia (see below).
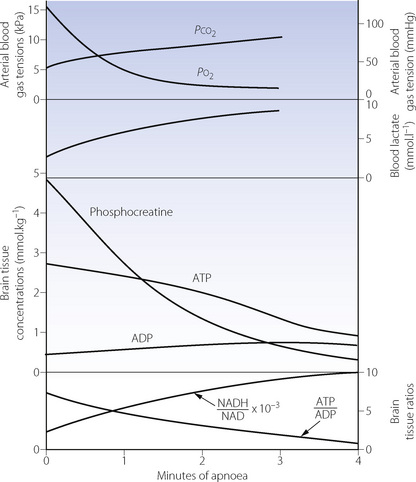
Under hypoxic conditions, there are two ways in which reductions in ATP levels may be minimised, both of which are effective for only a short time. First, the high energy phosphate bond in phosphocreatine may be used to create ATP,2 and initially this slows the rate of reduction of ATP (Figure 24.1). Secondly, two molecules of ADP may combine to form one of ATP and one of AMP (the adenylate kinase reaction). This reaction is driven forward by the removal of AMP (adenosine monophosphate), which is converted to adenosine (a potent vaso-dilator) and thence to inosine, hypoxanthine, xanthine and uric acid, with irreversible loss of adenine nucleotides. The implications for production of reactive oxygen species are discussed on page 383.
End-Products Of Metabolism
The end-products of aerobic metabolism are carbon dioxide and water, both of which are easily diffusible and lost from the body. The main anaerobic pathway produces hydrogen and lactate ions which, from most of the body, escape into the circulation, where they may be conveniently quantified in terms of the base deficit, excess lactate or lactate/pyruvate ratio. However, the blood–brain barrier is relatively impermeable to charged ions, and therefore hydrogen and lactate ions are retained within the neurones of the hypoxic brain. Lactacidosis can only occur when circulation is maintained to provide the large quantities of glucose required for conversion to lactic acid.
In severe cerebral hypoxia, a major part of the dysfunction and damage is due to intracellular acidosis rather than simply depletion of high energy compounds (see below). Gross hypoperfusion is more damaging than total ischaemia, because the latter limits glucose supply and therefore the formation of lactic acid. Similarly, patients who have an episode of cerebral ischaemia whilst hyperglycaemic (e.g. a stroke) have been found to have more severe brain injury than those with normal or low blood glucose levels at the time of the hypoxic event.3
Initiation of Glycolysis4
The enzyme 6-phosphofructokinase (PFK) is the rate-limiting step of the glycolytic pathway (see Figure 11.13). Activity of PFK is enhanced by the presence of ADP, AMP and phosphate, which will rapidly accumulate during hypoxia, thus accelerating glycolysis. PFK is, however, inhibited by acidosis, which will therefore quickly limit the formation of ATP from glucose. The intracellular production of phosphate from ATP breakdown also promotes the activity of glycogen phosphorylase, which cleaves glycogen molecules to produce fructose-1,6-diphosphate. This enters the glycolytic pathway below the rate-limiting PFK reaction, and also avoids the expenditure of two molecules of ATP in its derivation from glucose. Therefore four molecules of ATP are produced from one of fructose-1,6-diphosphate in comparison with two from one molecule of glucose. There is no subsequent stage in the glycolytic pathway that is significantly rate-limited by acidosis. Provided glycogen is available within the cell, this second pathway therefore provides a valuable reserve for the production of ATP.
Mechanisms of Hypoxic Cell Damage
Immediate Cellular Responses to Hypoxia5
Because of the dramatic clinical consequences of nervous system damage, neuronal cells are the most widely studied and therefore form the basis for the mechanisms described in this section.2 Changes in the transmembrane potential of a hypoxic neurone are shown in Figure 24.2, along with the major physiological changes that occur. At the onset of anoxia, CNS cells immediately become either slightly hyperpolarised (as shown in Figure 24.2) or depolarised, depending on the cell type. This is followed by a gradual reduction in membrane potential until a ‘threshold’ value is reached, when a spontaneous rapid depolarisation occurs. At this stage there are gross abnormalities in ion channel function and the normal intracellular and extracellular ionic gradients are abolished, leading to cell death.
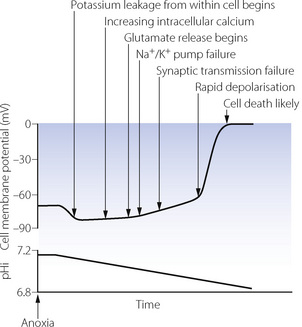
Potassium and sodium flux. Hypoxia has a direct effect on potassium channels (page 71), increasing transmembrane K+ conductance and causing the immediate hyperpolarisation. Potassium begins to leak out from the cell, increasing the extracellular K+ concentration thus tending to depolarise the cell membrane. Potassium leakage, along with sodium influx, are accelerated when falling ATP levels cause failure of the Na/K ATPase pump. Following rapid depolarisation, sodium and potassium channels probably simply remain open, allowing free passage of ions across the cell membrane leading to cellular destruction.
Calcium. Intracellular calcium concentration increases shortly after the onset of hypoxia. Voltage gated calcium channels open in response to the falling transmembrane potential and the increasing intracellular sodium concentration causes the membrane-bound Na/Ca exchanger to reverse its activity. An altered transmembrane potential is detected within the cell by ryonidine receptors on intracellular organelles leading to release of calcium from the endoplasmic reticulum and mitochondria.6
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


