Barry J. Maron, Iacopo Olivotto
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM), the most common of the genetic cardiovascular diseases, is caused by a multitude of mutations in genes encoding proteins of the cardiac sarcomere. HCM is characterized by heterogeneous clinical expression, unique pathophysiology, and diverse natural history.1–12 Although compatible with normal longevity in many patients, HCM represents the most common cause of sudden death in the young, including competitive athletes, and it is responsible for heart failure–related disability at virtually any age.1,3,8,11,13,14 Since the modern description of HCM more than 50 years ago,5,9,14 our understanding of the clinical complexity and spectrum of this disease has evolved dramatically.14 This chapter represents a contemporary and updated summary of HCM with respect to diagnosis, natural history, and management.
Definition, Prevalence, and Nomenclature
HCM is characterized by a thickened but nondilated left ventricle in the absence of another cardiac or systemic condition (e.g., aortic valve stenosis, systemic hypertension, some expressions of physiologic “athlete’s heart”) capable of producing the magnitude of left ventricular (LV) hypertrophy evident in this disorder1,3,11,15 (Figs. 66-1 and 66-2). Several epidemiologic studies have reported the prevalence of the HCM phenotype in the general population of approximately 0.2%, for a frequency of 1 in 500 people, equivalent to approximately 700,000 affected persons in the United States.1,11 This estimated frequency in the general population exceeds that implied by the relatively uncommon occurrence of HCM in cardiovascular practice, suggesting that most affected persons may remain unrecognized, usually without symptoms or cardiovascular events, during their lifetimes.1
HCM is a global disease reported in more than 50 countries from all continents, although most cases have come from the United States and Canada, Western Europe, Israel, and Asia (Japan and China).1 The first contemporary reports of HCM in 1958 were from Brock (in the cardiac catheterization laboratory) and from Teare, at autopsy describing “asymmetrical hypertrophy of the heart” as responsible for sudden death in a small group of young people. The disease rapidly acquired a confusing array of names, with most emphasizing the highly visible feature of LV outflow obstruction.15 Obstruction is not an invariable feature, however, and approximately one third of patients have the nonobstructive form of HCM; accordingly, names once in common use, such as idiopathic hypertrophic subaortic stenosis, hypertrophic obstructive cardiomyopathy, and muscular subaortic stenosis (as well as the acronyms, IHSS, HOCM, and MSS), have been largely abandoned. The preferred and generally accepted name for this condition is now hypertrophic cardiomyopathy (HCM) with or without outflow obstruction.
SEX and Race
As an autosomal dominant disease, HCM occurs with equal frequency in men and women.10,16–18 The predominance of men with HCM in the literature reflects underdiagnosis in women, who achieve clinical recognition less frequently, and at older ages, than men. Women are also at greater risk than men for progression to advanced heart failure (usually associated with outflow obstruction), although there is no relation between sex and sudden death risk or overall HCM-related mortality.19 HCM has been reported in many races, but appears to be under-recognized in African Americans, and most competitive athletes who die suddenly of HCM are previously undiagnosed black men.13 Clinical expression, presentation and course of HCM, as well as phenotypic genetic expression are similar throughout the world, although the morphologic form characterized by hypertrophy confined to the LV apex is more prevalent in Japan.
Genetic Basis and Laboratory Testing
HCM is transmitted as a mendelian trait with an autosomal dominant pattern of inheritance (see also Chapter 8); every offspring of an affected relative has a 50% chance of developing the disease.4,10,16–18,20 Molecular studies, conducted intensively over two decades, have provided access to definitive laboratory-based diagnosis by identifying disease-causing mutations, and providing in the process important insights into the broad clinical expression of HCM, including that in persons carrying pathogenic mutations without evidence of the disease phenotype.4,16–18,20
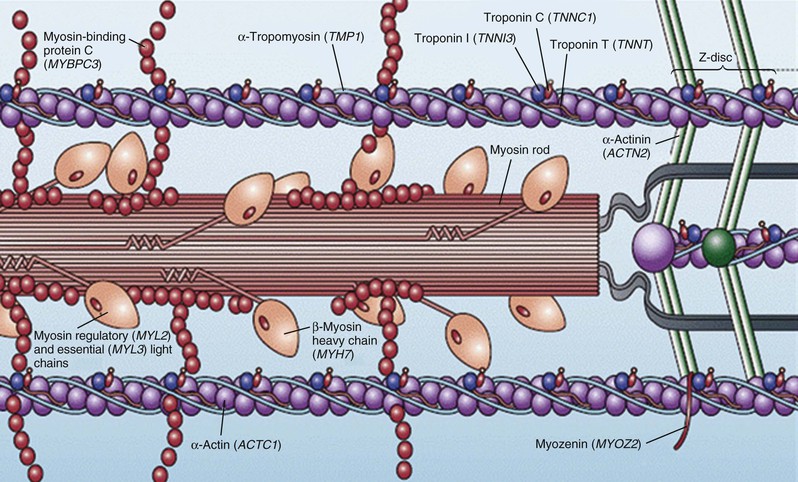
HCM is now known to be caused by mutations in 11 or more genes encoding proteins of the thick and thin contractile myofilament components of the cardiac sarcomere or the adjacent Z-disc4 (Figs. 66-3 to 66-5). Two sarcomere genes, those encoding β-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3), are by far the most common, accounting for 30% of consecutively screened patients with HCM and 70% of those successfully genotyped. The troponin T gene (TNNT2) and several others are each responsible for up to 5% of cases. Underscoring the vast genetic heterogeneity of HCM is the recognition of about 1500 individual mutations (largely missense) identified in patients, most of which are unique to individual families4 (see Fig. 66-4).
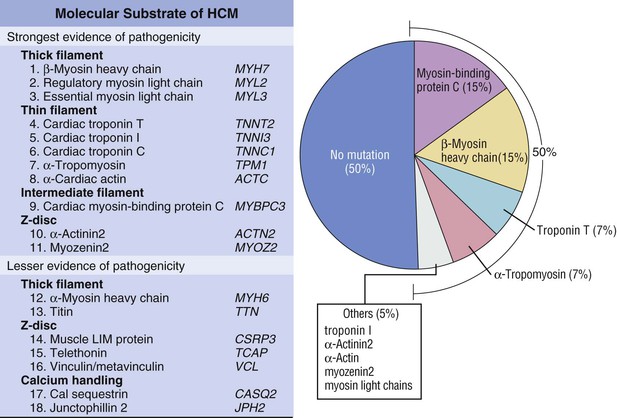
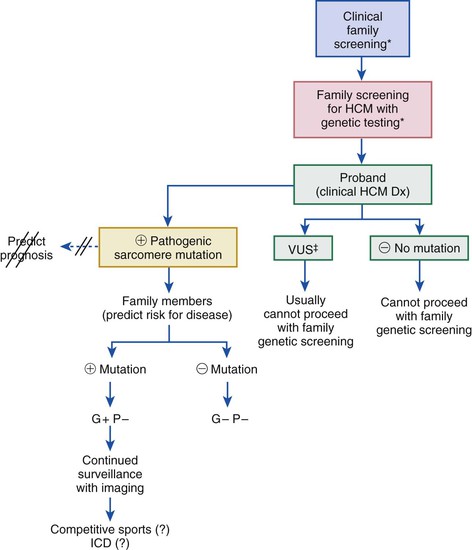
Rapidly automated DNA sequencing now provides opportunities for comprehensive commercially available genetic testing (see Fig. 66-5). To date, the greatest power of molecular testing currently lies with the capability to identify or exclude affected status in family members without LV hypertrophy.4,10,16 This strategy first requires identification of a pathogenic mutation in a relative (proband) with clinically expressed HCM. Pathogenic mutations are recognized in less than 50% of clinically affected probands, however, and DNA-based testing not infrequently identifies novel but ambiguous sequence variants for which pathogenicity is unresolved (variants of unknown significance).4 Such variants have no clear cut application to clinical family screening, underscoring the challenges remaining in translating complex molecular science to patient care. The possibilities offered by next generation techniques may enhance screening capabilities and reduce costs, but also will increase the number of variants of unknown significance.
Of note, despite early promising genetic data, predicting prognosis and risk for sudden death in individual patients with HCM based on specific sarcomere mutations has proved to be unreliable.4,8,11,18,20 Some evidence indicates that multiple mutations coexisting in the same patient lead to early disease onset and severe clinical profile.4 Genetic testing can, however, clarify diagnosis for patients with metabolic and storage disorders in which clinical presentation and pattern of LV hypertrophy mimic those in sarcomeric HCM, but in which pathophysiology, natural history, and management are dissimilar, including Fabry disease, PRKAG2, and LAMP2 (Danon’s disease).21 Such genetic diagnoses are of substantial clinical value. For example, LAMP2 cardiomyopathy is associated with a lethal natural history refractory to defibrillation therapy (with survival uncommon beyond 25 years), necessitating early recognition and heart transplantation,21 whereas enzyme replacement therapy is available for patients with Fabry disease.
Morphology and Role of Cardiac Imaging
Hypertrophic Cardiomyopathy Phenotype/Left Ventricular Hypertrophy
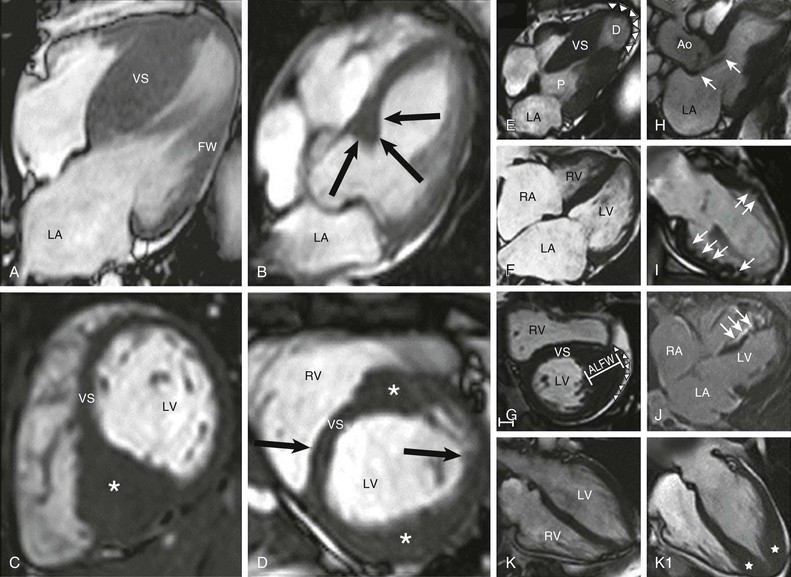
The clinical diagnosis of HCM is conventionally made by imaging with two-dimensional echocardiography. However, cardiovascular magnetic resonance (CMR) (see also Chapter 17) has more recently emerged with an expanded role in noninvasive diagnosis by virtue of its high-resolution tomographic imaging capability5 (see Fig. 66-2). Furthermore, CMR allows for the quantification of late gadolinium enhancement, which is a marker of myocardial fibrosis potentially relevant to risk stratification in HCM, as well as identification of disease progression to the end-stage phase.6,22–25 In HCM, CMR imaging is complementary to echocardiography by clarifying technically ambiguous LV wall thicknesses, or visualizing clinically relevant abnormalities that may clarify diagnosis or alter management strategies not reliably identified with echocardiography. These include areas of segmental hypertrophy in the anterolateral free wall or posterior portion of septum,26 or pathology in the apical LV chamber such as hypertrophy and aneurysm formation.27 Cardiac imaging in clinically identified adults and children typically documents absolute increase in LV wall thickness (21 to 22 mm on average, and up to >50 mm), although no particular wall thickness is inconsistent with genetically affected status. Diverse patterns of asymmetric LV hypertrophy are characteristic of HCM (see Fig. 66-2), even within the same family (with the exception of identical twins). Typically, one or more regions of LV chamber are of greater thickness than other areas, often with sharp transition in thickness between adjacent areas or noncontiguous patterns of segmental hypertrophy (Fig. 66-2D), as well as extension into the right ventricle in some patients.28 No single morphologic form of HCM is considered “classic” or typical, and none are consistently related to outcome.5,25
Mitral Valve Apparatus
Primary structural abnormalities of the mitral apparatus responsible for LV outflow obstruction are part of HCM phenotypic expression (see Fig. 66-2H). The mitral valve may be more than twice normal size as a consequence of elongation of both leaflets, or segmental enlargement of only the anterior leaflet or mid-scallop of the posterior leaflet.37 Furthermore, congenital and anomalous anterolateral papillary muscle insertion directly into anterior mitral leaflet (without interposition of chordae tendineae) may occur.5
Histopathology
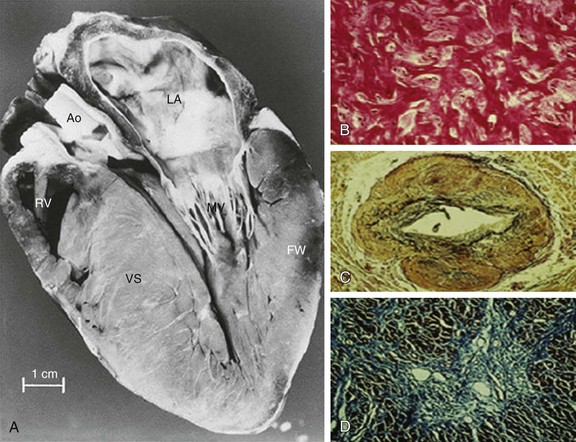
In HCM, hypertrophied cardiac muscle cells (myocytes) in both ventricular septum and LV free wall exhibit bizarre shapes, often maintaining intercellular connections with several adjacent cells.1 Many myocytes (and also myofilaments) are arranged in chaotic and disorganized architectural patterns (Fig. 66-1B). Areas of cell disarray are evident in 95% of HCM hearts at autopsy, usually occupying substantial portions of hypertrophied (as well as nonhypertrophied) LV myocardium (i.e., 33% of septum and 25% of free wall).
At autopsy, the vast majority of patients with HCM exhibit intramural coronary arterioles with marked thickening of the vessel wall secondary to smooth muscle hyperplasia in the media (associated with abundant disorganized elastic fibers), causing deformation and narrowing of the lumen (Fig. 66-1C), and frequently located within or close to areas of myocardial scarring. This microvascular small-vessel disease probably is responsible for repeated bursts of clinically silent myocardial ischemia leading to myocyte death, with a repair process in the form of replacement (and often transmural) myocardial fibrosis (Fig. 66-1D).38 In addition, volume of the interstitial (matrix) collagen compartment, constituting the structural LV framework, is greatly expanded.
It is likely that the interplay of disorganized cellular architecture, microvascular ischemia, and replacement fibrosis (and microvascular ischemia) impairs transmission of electrophysiologic impulses and predisposes to disordered patterns and increased dispersion of electrical depolarization and repolarization. This, in turn, serves as an electrophysiologically unstable substrate and a trigger for reentry ventricular tachyarrhythmias and sudden death.
Pathophysiology
Left Ventricular Outflow Tract Obstruction
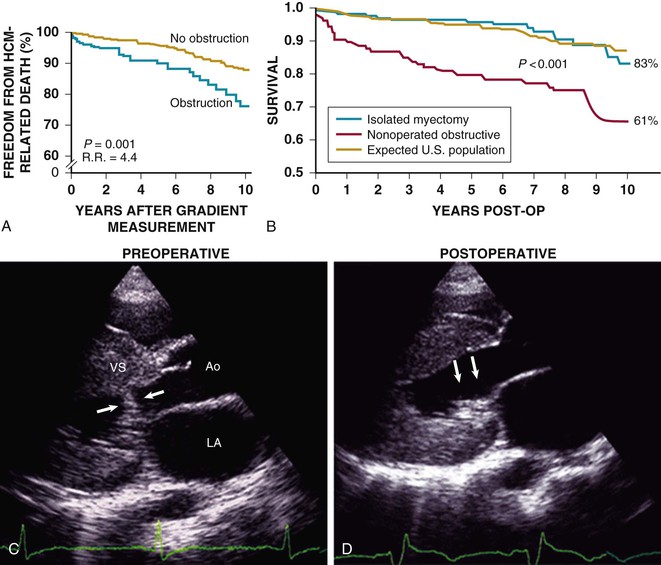
HCM is predominantly an obstructive disease in that fully 70% of patients show the propensity to develop dynamic LV outflow gradients of 30 mm Hg or greater, either at rest or during exercise: the true nonobstructive form accounts for approximately one third of cases.1,7,9 Longstanding outflow obstruction is a strong determinant of HCM-related progressive heart failure symptoms and cardiovascular death (Figs. 66-6 and 66-7).1,9,39 However, only a weak relationship is evident between outflow obstruction and risk for sudden death.
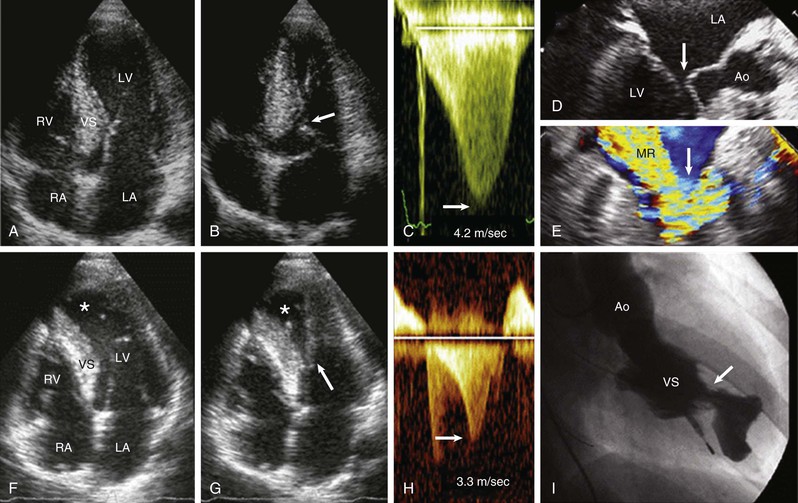
Subaortic obstruction in HCM represents true mechanical impedance to LV outflow, producing markedly increased intraventricular pressures that over time may be detrimental to LV function, probably by increasing myocardial wall stress and oxygen demand (see Fig. 66-6). In the vast majority of patients, obstruction is produced in the proximal LV by systolic anterior motion (SAM) of the mitral valve in which elongated leaflets bend sharply at 90 degrees and contact the septum in midsystole owing to a drag effect—that is, hydrodynamic pushing force of flow directly on the leaflets (Fig. 66-6A-E). The magnitude of the outflow gradient, which is reliably estimated with continuous wave Doppler, is directly related to the duration of mitral valve–septal contact. Mitral regurgitation is a secondary consequence of SAM, with the jet (usually mild to moderate in degree) directed posteriorly (Fig. 66-6E). Marked and centrally located mitral regurgitation jets usually suggest an intrinsic valve abnormality (e.g., with myxomatous degeneration). Occasionally, intraventricular obstruction may occur at mid-cavity level caused by systolic contact of septum with a papillary muscle that is anomalously positioned and may insert directly into anterior mitral leaflet (Fig. 66-6F-I). This form of obstruction may be associated with LV apical aneurysm.
Subaortic gradients (and related systolic ejection murmurs) in HCM are often dynamic, with spontaneous variability, and are reduced or abolished by interventions that decrease myocardial contractility (e.g., beta-adrenergic blocking drugs) or increase ventricular volume or arterial pressure (e.g., squatting, isometric handgrip, phenylephrine). Alternatively, gradients can be augmented by circumstances in which arterial pressure or ventricular volume is reduced (e.g., Valsalva maneuver, administration of nitroglycerin or amyl nitrite, blood loss, dehydration) or when LV contractility is increased, such as with premature ventricular contractions, infusion of isoproterenol or dobutamine, or with exercise. Consumption of a heavy meal or alcohol can also transiently increase subaortic gradients and produce exertional dyspnea. Notably, a large proportion of patients without outflow obstruction (or SAM) at rest may generate outflow gradients with physiologic exercise, which are sometimes associated with severe heart failure symptoms, but which can be blunted by inhibition of sympathetic stimulation with beta blockers.40
Diastolic Dysfunction
Evidence of impaired LV relaxation and filling, by pulsed and tissue Doppler imaging,41 is present in most patients with HCM, probably contributing to symptoms of exertional dyspnea, although unrelated to severity of LV hypertrophy.1 Diastolic dysfunction (see also Chapters 14 and 27) is the likely cause of limiting symptoms in patients with nonobstructive disease, and represents the mechanism by which progressive heart failure may occur despite preserved LV systolic function. Such patients occasionally become refractory to medical management, requiring heart transplantation.1
Reduced ventricular compliance in HCM probably results largely from those factors determining passive elastic properties of the LV chamber, such as hypertrophy, replacement scarring, interstitial fibrosis, and disorganized cellular architecture. Furthermore, abnormal energetic handling and diastolic calcium overload occurring at the cardiomyocyte level actively contribute to diastolic dysfunction. The most commonly observed pattern in HCM is delayed relaxation, characterized by a prolonged rapid filling phase associated with decreased rate and volume of LV filling and (in sinus rhythm) a compensatory increase in the contribution of atrial systole to overall filling.41 However, most echocardiographic measurements of diastolic dysfunction do not reliably predict prognosis, symptoms, or filling pressures, with consequent limited clinical application to management of patients with HCM, although restrictive filling patterns may have adverse prognostic implications.41,42
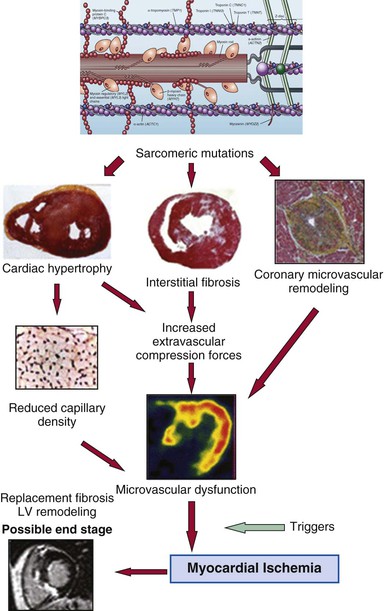
Microvascular Dysfunction
Myocardial ischemia due to microvascular dysfunction appears to be an important pathophysiologic component of the HCM disease process, promoting adverse LV remodeling and ultimately affecting clinical course38,43 (Fig. 66-8). Marked reduction in coronary reserve can be demonstrated in patients with HCM using positron emission tomography (PET) early in the clinical course (in both hypertrophied and nonhypertrophied regions of the left ventricle) and is an important determinant of prognosis, including progressive heart failure, systolic dysfunction, and long-term cardiovascular mortality. PET represents the technique of choice for the assessment of microvascular function in patients with HCM,43 although its systematic use has not entered clinical practice systematically (see also Chapter 16).