Hypertrophic cardiomyopathy (HCM) is a heart muscle disease that results in cardiac hypertrophy and oftentimes left ventricular outflow tract (LVOT) obstruction. Both the structural and hemodynamic alterations impart risk of arrhythmia, heart failure, and sudden death in those affected. In >50% of patients, causative mutations in genes that encode proteins related to the structure and function of the sarcomere are found.
HCM is characterized by ventricular hypertrophy in the absence of ventricular dilatation in initial stages. The hypertrophy often shows a predilection for the ventricular septum at the base of the heart, resulting in asymmetric (nonconcentric) thickening, though midventricular, apical, and diffuse (neutral) forms can also be seen. Concentric hypertrophy can also occur.1
The septal hypertrophy results in LVOT obstruction, previously called “idiopathic hypertrophic subaortic stenosis” and currently designated “hypertrophic obstructive cardiomyopathy” (HOCM). Obstruction to LVOT is caused, in part, by bulging of the thickened septum. This altered LVOT geometry causes abnormal drag forces to be applied to the anterior mitral leaflet (posterior portion of the LVOT) and causes it to move anteriorly, which can further obstruct blood from ejection into the aorta. The designation HCM should be used for all morphologic variants, however, as the pathogenesis and genetics are similar.
HCM is most often inherited in an autosomal dominant fashion with variable and commonly incomplete penetrance. Symptoms and presentation vary substantially between cohorts and can range from in utero presentation to late adulthood.2,3 Because patients often present initially as sudden death, typically during exertion, HCM is one of the best known cardiomyopathies.
Epidemiology
Defined by echocardiographic measurements, HCM affects about 1:500 of the general population.4 With molecular genetic screening, some believe the rate may be higher, although most data show a similar rate to that seen with imaging.5,6 Relatives of probands have some morphologic evidence of the disease in up to 25% of cases. HCM most commonly presents in the second or third decade of life but may present at any age. It is the most common identified substrate for sudden unexpected death among young athletes, although it is significantly more common to find nothing at all by conventional autopsy.7 The incidence is low, however, in screening studies of competitive athletes.8
Clinical Features and Diagnosis
HCM is diagnosed in three groups of patients. First are patients who present with symptoms that are nonspecific and include dyspnea, especially during exercise; chest pain; palpitations; dizziness; and syncope. This first group is commonly young in the 2nd to 4th decades of life, and the diagnosis is confirmed by echocardiography and other imaging techniques.9,10 The second and largest group is composed of either relatives of patients with the disease or patients who are found to have HCM incidentally. These patients are either asymptomatic or minimally symptomatic. The third group consists primarily of those who die suddenly without a prior history. In this group, the pathologist has a key role in establishing the diagnosis and establishing a basis for genetic counseling for family members.
Echocardiography
HCM is diagnosed by unexplained left ventricular hypertrophy, small left ventricular cavity size, and increased or normal left ventricular systolic function. Left ventricular hypertrophy may be either nonconcentric or concentric, with the former being more specific for the diagnosis than the latter. On echocardiography, a left ventricular thickness of more than 15 mm is suspicious for the disease. Tissue Doppler imaging has been recently applied to obtain an early diagnosis of HCM, making echocardiography, including Doppler evaluation, the most commonly used diagnostic technique.11
There are several conditions that may mimic the imaging features, especially amyloidosis and Fabry disease. Over 5% of patients over 40 years referred for the treatment of HCM are found to actually suffer from Fabry disease after serum levels of serum alpha-galactosidase are measured.12,13 Additionally, the preferential deposition of amyloid in the basal ventricular septum can mimic the asymmetric hypertrophy seen in HCM.
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging is useful both diagnostically and prognostically in the assessment of patients with suspected HCM.14 The presence of myocardial scar detected by late gadolinium enhancement (LGE) on cardiac magnetic resonance (CMR) imaging has been described as a good independent predictor of mortality and the risk of sustained ventricular tachycardia in patients with HCM.15,16
Arrhythmias in HCM
The prognosis of HCM is based primarily on the propensity for ventricular tachyarrhythmias and secondarily based on the development of late heart failure. A history of syncope is associated with a higher incidence of sudden death, and patients with a significant LVOT obstruction are more susceptible to clinical deterioration.17,18 Risk stratification to evaluate the need for implantable defibrillation is based on clinical and electrocardiographic features.19 The degree of ventricular hypertrophy or outflow obstruction is not particularly helpful, as certain genotypes (e.g., troponin mutations) are associated with arrhythmias in the absence of marked cardiomegaly.
Atrial fibrillation occurs in up to 20% of patients with HCM and is associated with older age, symptomatic disease, and decreased overall survival, but not sudden death. Radiofrequency ablation for atrial fibrillation may be effective in some patients.20,21
Genetics
More than half of patients with HCM have a family history of the disease, and the pattern of inheritance is usually autosomal dominant. In 1990, a seminal work led to the identification of a causal gene and mutation for HCM, a missense mutation in the cardiac β-myosin heavy chain gene (MYH7).22
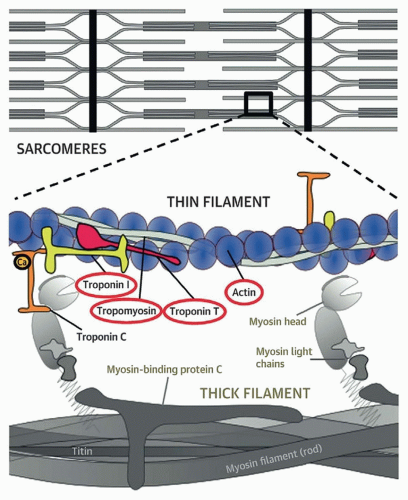
Since then, multiple genes have been identified in HCM patients. Dominant pathogenic variants in HCM genes include sarcomeric genes that involve thick filaments (myosin-binding protein C [MYBPC3], β-myosin heavy chain [MYH7], and myosin light chains 2 and 3 [MYL2 and MYL3]) and thin filaments (actinin1 [ACTC1], cardiac troponin I type 3 [TNNI3], cardiac troponin T type 2 [TNNT2], and tropomyosin-1 [TPM1]) (Table 157.1; Fig. 157.1). Additional genes, including ACTN2, CSRP3, MYOZ2, NEXN, PLN, TNNC1, and TTN, have been implicated but not definitively proven as causing HCM.
TABLE 157.1 Prevalence of Mutations in Hypertrophic Cardiomyopathy and Their Respective Clinical Characteristics
Gene
Characteristics
Frequency
Myosin-binding protein C (MYBPC3)
Incomplete and late penetrance, relatively benign course
Additional genes, including ACTN2, CSRP3, MYOZ2, NEXN, PLN, TNNC1, and TTN, have been implicated but not definitively proven as causing disease. Pathogenic variants in other genes (GLA/Fabry disease, LAMP2/Danon cardiomyopathy, and PRKAG2/glycogenosis) also cause left ventricular hypertrophy but result in metabolic disorders distinct in their origin from HCM.23
aMYH6 and MYH7 are isoforms of the myosin heavy chain, the latter primarily expressed in the myocardium. There is a high degree of homology between them, so most studies do nottest for MYH6 separately. Recently, a high rate of MYH6-specific mutations confirmed by sequencing was found in a series of HCM.10
Other gene mutations in GLA, LAMP2, and PRKAG2 cause left ventricular hypertrophy that may mimic HCM but are distinct disorders (Fabry disease, Danon cardiomyopathy, and PRKAG2-associated cardiomyopathy, respectively, the latter generally classified as a glycogenosis) (see Chapter 23).23 Noonan syndrome, associated with mutations in a number of genes, PTPN11, SOS1, and RAF1 being the most common, is also usually associated with cardiomegaly that mimics HCM. Importantly, these should NOT be considered HCM but rather heart disease associated with those conditions.
FIGURE 157.1 ▲ Genes associated with the pathogenesis of hypertrophic cardiomyopathy (HCM). Genes encoding proteins in the thick filament that cause hypertrophic cardiomyopathy include myosin-binding protein C and myosin heavy chain. Mutations in titin are associated primarily with dilated cardiomyopathy. Genes encoding proteins in the thin filament associated with HCM include troponins, tropomyosin, and actin (circled). Less commonly, HCM is caused by mutations in myosin light chains and actinin (not shown, involved in the attachment of actin filaments to the Z-line). (Reproduced with permission, Coppini R, Ho CY, Ashley E, et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin filament gene mutations. J Am Coll Cardiol. 2014;64:2589-2600.)
The detection rate of causative mutations in unselected probands with HCM varies, depending on the method used and definition of causation. The rate ranges from 32%23 to 46%24 and 64%.10
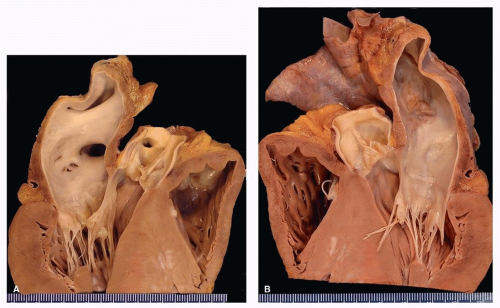
FIGURE 157.2 ▲ Hypertrophic cardiomyopathy. A. Long-axis section, base of heart, anterior left side of heart. There is thickening of the ventricular septum with focal gross scars. No outflow tract plaque is present in this case. B. Long-axis section. There is left atrial dilatation, typical of the disease, secondary to stiffening of the ventricle. There is no ventricular dilatation in early stages of disease. (Reprinted from Tavora F, Cresswell N, Li L, et al. Morphologic features of exertional versus nonexertional sudden death in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2010;105:532-537, with permission.)
Mutations are classified as pathogenic, likely pathogenic, likely benign, benign, or of undetermined significance. This classification is based on frequency of the observed variant, functional studies, and computer-based prediction tools. These variants can be further categorized by the effect they have on the RNA transcript: missense, nonsense, insertions, deletions, or splice site.23 The art and science of these genetic variant cells are ever improving and will likely have significant impact on the understanding of this disease over the next decade.
Only gold members can continue reading. Log In or Register to continue