Fig. 11.1
12-lead ECG
The initial EKG showed sinus rhythm at 58 bpm, normal atrioventricular conduction (PR interval 160 s), normal QRS duration (0.08 s), absent Q wave, and horizontal QRS axis (+15°). Increased amplitude and duration (>0.04 s) of the terminal negative portion of P wave in V1. High-voltage QRS complexes. T-wave inversion in the left precordial leads, reciprocal ST-segment elevation, and tall T wave in the right precordial leads. ST-segment depression and T-wave inversion in lead I and aVL.
The Sokolow diagnostic criteria for left ventricular hypertrophy (LVH) were satisfied: R wave in V6 + S wave in V1 >3.5 mV.
Asymmetrical configuration of inverted T waves suggested a nonischemic origin.
Conclusion: sinus rhythm, normal conduction, possible left atrial enlargement, and left ventricular hypertrophy with secondary anomalies of repolarization
We hypothesized that the described palpitations were provoked by a hyperkinetic arrhythmia (supraventricular or ventricular), and we explored the different secondary causes of arrhythmic events:
Hyperthyroidism
Fever
Anxiety
Anemia
Use of medications containing stimulant, caffeine, or nicotine
Strenuous exercise, poor training
He did not report any specific stress condition or recent changes in his lifestyle. He was walking slowly, when the arrhythmia occurred, which therefore can be excluded a physiologic activity response and poor training. According to physical examination, fever was excluded, and the laboratory tests did not show any anemia and thyroid dysfunction.
The most probable cause of symptoms was spontaneous arrhythmia.
Transthoracic Echocardiography (TTE)
A TTE showed (Figs. 11.2, 11.3, and 11.4):
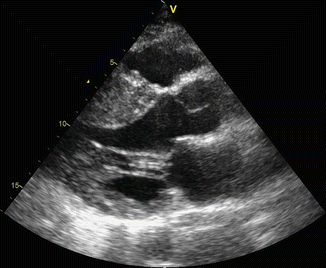
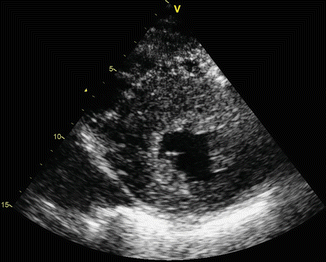
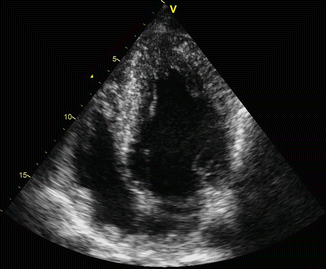
Fig. 11.2
Parasternal long-axis view shows septal hypertrophy
Fig. 11.3
Parasternal short-axis ventricle view
Fig. 11.4
Apical four-chamber view
Left ventricular volumes at lower normal limits (iLVEDV 35 mL/m2) with massive asymmetrical hypertrophy with septal wall thickness that reached 35 mm; the posterior wall was 15 mm with a septum to posterior wall ratio of 2.4.
Ejection fraction with the biplane Simpson method was 65 % without regional wall motion abnormalities.
Diastolic dysfunction grade II with normal estimated filling pressure (E/E’ 6).
Severe left atrial enlargement (LA diameter 55 mm, iLAV >40 mL/m2).
Systolic anterior motion (SAM) of the anterior mitral valve leaflet, without LVOT obstruction at rest, but with mild flow acceleration during Valsalva (peak gradient 15 mmHg).
Normal size and function of right atrium and ventricle.
Normal aortic tricuspid valve with mild central regurgitation
Normal tricuspid and pulmonic valves.
Normal dimension of inferior vena cava (IVC) with >50 % inspiratory collapse.
No pericardial effusion.
According to ESC guidelines, HCM is defined as a wall thickness >15 mm in one or more LV myocardial segments, as measured by any imaging technique (echocardiography, cardiac magnetic resonance imaging (CMR), or computed tomography (TC)), that is not explained solely by chronic loading conditions.
Echocardiographic findings were suggestive of hypertrophic cardiomyopathy: the patient had no history of hypertension or valve disease with elevated afterload. Infiltrative cardiomyopathy was excluded, because the ventricular thickening is usually concentric with characteristic pattern of granular sparkling and different degrees of pericardial effusion.
In a patient with HCM, a sustained episode of palpitation lasting more than few minutes is often caused by supraventricular arrhythmias, especially in the presence of left atrium enlargement; atrial fibrillation is the most common arrhythmia in this population. In our patient, we could not exclude the hypothesis of a ventricular origin, particularly because the symptoms associated with palpitation (dyspnea and dizziness) could point to a hemodynamic distress that is often related to sustained ventricular tachycardias.
In adult patients with HCM, most recent data report on an annual incidence of cardiovascular death near 1–2 % with sudden cardiac death being (SCD) the most common.
Major clinical features associated with an increased risk of SCD are:
Young age
Non-sustained ventricular tachycardia (NSVT)
Maximum left ventricular wall thickness
Family history of sudden cardiac death
Syncope
Left atrial diameter
Left ventricular outflow tract obstruction
Exercise blood pressure drop
The patient had no family history of SCD and denied syncope, but the risk assessment comprised also of a 24-h ambulatory ECG and an exercise test.
Exercise Testing with Treadmill
Exercise testing was terminated for asthenia and muscular weakness at a heart rate corresponding to 93 % of maximal heart rate predicted for age. Neither symptoms nor ST-segment depression or elevation occurred. During exercise, arrhythmias were not detected except for two isolated polymorphic ventricular ectopic beats (VE) and one VE couple. Systolic blood pressure and heart rate response were normal.
24-h Ambulatory ECG
24-h ambulatory ECG was performed to detect atrial or ventricular arrhythmias. The total number of beats analyzed was 82,105. Sinus rhythm at average heart rate of 64 bpm, with minimum of 50 bpm and a maximum of 95 bpm. Two hundred fifty total ventricular ectopic (VE) beats of different morphologies with six VE couples and one NSVT (four beats at 150 bpm). Forty-three total supraventricular ectopic beats with one short run of 12 beats. No significant pauses.
NSVT is defined as ≥3 consecutive ventricular beats at ≥120 bpm lasting <30 s, so only one episode was detected.
In the absence of sustained arrhythmia, the electrophysiological study (EPS) is not specifically recommended.
Clinical Course
According to HCM guidelines, ICD implantation should be considered in patients with an estimated risk of SCD ≥6 % and a life expectancy of >1 year, and it may be considered in patients with an estimated risk between ≥4 and <6 %, while it is not recommended in patients with an estimated risk <4 % unless they have clinical features that are of proven prognostic importance.
Our patient’s estimated risk of sudden cardiac death at 5 years was 5 % (intermediate), based on severe cardiac hypertrophy and NSVT; that supported the indication for ICD implant in primary prevention.
The patient was informed about his SCD risk and the necessity of an ICD implant. He was made also aware on the risk of inappropriate shocks, implant complications, and the social and occupational implications of an ICD implant.
According to young age, primary prevention indication, good AV conduction, and patient’s preference, we scheduled a subcutaneous ICD (S-ICD) implantation.
At the preimplantation screening test, the patient presented the anatomical and electrocardiographic features ideal for a suitable subcutaneous sensing. An S-ICD was then implanted without complications.
Chest X-Ray
Chest X-ray was performed the day after implantation (Figs. 11.5 and 11.6).
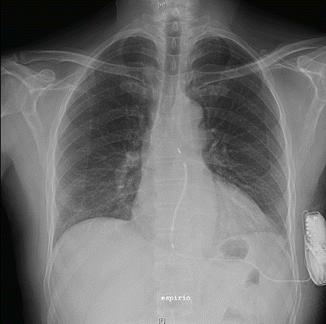
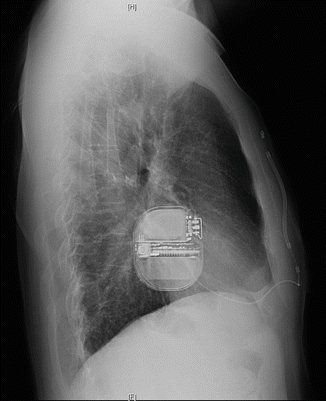
Fig. 11.5
Chest X-ray anterior-posterior (AP) projection
Fig. 11.6
Chest X-ray lateral view
Conclusion: Correct position of the implanted S-ICD system
Therapy and Discharge
Because the episode of prolonged palpitation was his first and an S-ICD was implanted, we decided not to give antiarrhythmic therapy and maintained the calcium channel blockers (verapamil) in order to control LVOT gradient.
Moreover, at discharge, the patient was advised to abstain from competitive athletic activity and strenuous physical exertion and was given clinical and echocardiographic follow-up appointments.
11.2 Hypertrophic Cardiomyopathy
Introduction and Epidemiology
The most recent expert consensus on cardiomyopathies has adopted a new classification system no more based on primary or secondary involvement of the heart but in which cardiomyopathies are defined by specific morphological and functional phenotypes as they present for the first time to the observer: mainly hypertrophic, dilated, arrhythmogenic cardiomyopathy and restrictive phenotype. Only in the second time, cardiomyopathies are grouped into familial/genetic and nonfamilial/nongenetic subtypes, irrespective of the presence of extra-cardiac disease [1, 2, 3].
Hypertrophic cardiomyopathy (HCM) is defined by the presence of increased left ventricular (LV) wall thickness that is not solely explained by abnormal loading conditions. Many are the secondary causes of hypertrophy (in particular left ventricular hypertrophy) that should be considered in differential diagnosis:
1.
Athlete’s heart
2.
Hypertensive cardiomyopathy
3.
Valve diseases imposing increased afterload (mainly aortic stenosis)
4.
Isolated basal septal hypertrophy in elderly people
Moreover hypertrophic phenotype (variable grade and distribution of ventricular wall thickening) could represent a common picture of different pathologic conditions such as infiltrative disorders due to inborn errors of metabolism (e.g., Pompe disease, Fabry disease) or deposition of anomalous misfolded proteins (different types of amyloidosis). Other genetic causes could be mithocondrial diseases, neuromuscular disorders (Friedreich’s ataxia), or malformative syndromes like Noonan or LEOPARD [4].
The true hypertrophic cardiomyopathy is a genetic disease with an autosomal dominant trait caused by mutations in cardiac sarcomere protein genes. In general, patients with a sarcomere protein mutation present earlier and report a higher prevalence of family history of HCM and sudden cardiac death (SCD) than those without a mutation. They also tend to have more severe hypertrophy, microvascular dysfunction, and myocardial fibrosis.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


