Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is the most common genetic cardiovascular disease.1 The prevalence in the general adult population for people with phenotypic evidence of HCM is estimated at 1 per 500.2 It is the most common etiology for sudden cardiac death (SCD) in young adults.3,4
HCM has traditionally been defined as myocardial hypertrophy of ≥1.5 cm without an identifiable cause (Figs. 22.1 and 22.2). Other etiologies of hypertrophy must be excluded before diagnosing HCM (Table 22.1). While there are multiple synonyms for HCM, including muscular subaortic stenosis (MSS), hypertrophic obstructive cardio-myopathy (HOCM), and idiopathic hypertrophic subaortic stenosis (IHSS), the World Health Organization (WHO) recommends that HCM be used as the term for the disease.
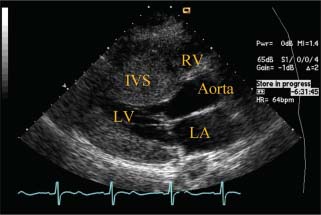
FIGURE 22.1 Transthoracic echocardiogram of HCM in the young—diffuse hypertrophy. Parasternal long-axis view depicts a markedly thickened interventricular septum. The thickening is diffuse, extending from base to beyond the midventricle. LV, left ventricle; RV, right ventricle; LA, left atrium; IVS, interventricular septum.
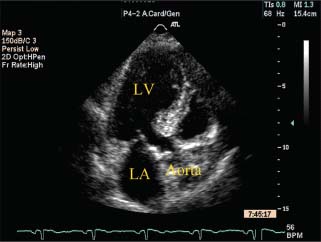
FIGURE 22.2 Transthoracic echocardiogram of HCM in the elderly—proximal septal hypertrophy. Apical three-chamber view depicts focal thickening of the interventricular septum at its base. The midventricle appears to be uninvolved. LV, left ventricle; LA, left atrium.
TABLE
22.1 Alternative Causes of LV Wall Thickening

CLASSIFICATION
HCM can be classified as obstructive or nonobstructive, depending on the presence of a left ventricular outflow tract (LVOT) gradient, either at rest or with provocative maneuvers. Seventy percent of subjects with HCM have LVOT gradients ≥30 mm Hg at rest or with exercise.5 Obstruction is caused by systolic anterior motion (SAM) of the mitral valve leaflet.
Anatomic variants of HCM exist, and these can be categorized based on the location of the hypertrophy (e.g., proximal septal, apical, or diffuse). Apical hypertrophy is also known as Yamaguchi disease (Fig. 22.3). Additionally, distinct forms of HCM appear to exist, depending on age. Younger patients tend to have hypertrophy of the entire septum (see Fig. 22.1), whereas older patients generally have basal septal hypertrophy, known as a sigmoid septum (see Fig. 22.2).6 It is believed that these are two different disease processes. The majority of elderly HCM (diagnosed at >50 years of age) were negative for mutations for HCM, especially when a sigmoid septum was present, whereas younger subjects with HCM were more likely to have mutations for HCM.7
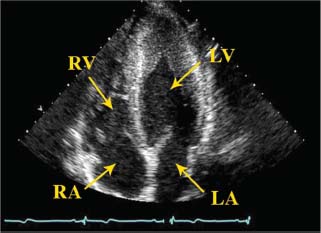
FIGURE 22.3 Transthoracic echocardiogram of apical variant of HCM (Yamaguchi). Apical four-chamber view depicts ventricular thickening of the apex. LV, left ventricle; LA, left atrium; RV, right ventricle; RA, right atrium.
PATHOPHYSIOLOGY AND HISTOLOGY
LVOT obstruction is the pathophysiologic change in obstructive HCM (Figs. 22.4 and 22.5). When SAM occurs, the mitral valve leaflets are pulled or dragged anteriorly toward the ventricular septum, producing LVOT obstruction.8 The left ventricle (LV) thus must generate higher pressures to overcome the obstruction and to pump blood systemically. Premature closure of the aortic valve frequently occurs, caused by the decline in pressure distal to the LVOT obstruction.
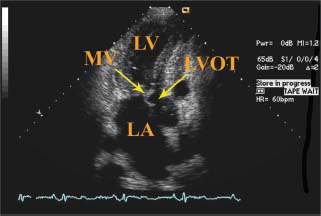
FIGURE 22.4 Transthoracic echocardiogram of SAM of mitral valve. Apical three-chamber view illustrates SAM of the mitral valve, resulting in obstruction of the LVOT. LV, left ventricle; LA, left atrium; MV, mitral valve; LVOT, left ventricular outflow tract.
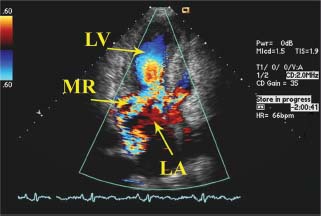
FIGURE 22.5 Transthoracic echocardiogram of mitral regurgitation in HCM. Apical three-chamber view illustrates the classic posterolaterally directed mitral regurgitation jet in HCM. The jet direction occurs secondary to SAM of the mitral valve. LV, left ventricle; LA, left atrium; MR, mitral regurgitation jet.
Dynamic obstruction occurs with HCM, whereas fixed obstruction occurs with aortic stenosis and subvalvular aortic membranes. In dynamic obstruction, the degree of obstruction depends to a larger extent on cardiac contractility and loading conditions. This is contrasted in fixed obstruction, where cardiac contractility and preload have little effect on the degree of obstruction. An underfilled LV results in greater obstruction because there is less separation between the interventricular septum and the mitral valve. As the LV cavity gets smaller and the flow stream is directed against the mitral valve, the SAM of the mitral valve occurs. Augmenting cardiac contractility also increases LVOT obstruction, because a more vigorous contraction is more likely to cause the obstructing components to come into contact. As the mitral leaflet comes closer against the septum, the outflow tract is decreased in size, which further increases the pressure difference. This feedback loop is represented on continuous-wave Doppler imaging as the concave contour (Fig. 22.10).
Histologically, HCM manifests as hypertrophied, disorganized cardiac myocytes present throughout the myocardium. The abnormal cells may take on bizarre shapes, and the connections among cells are often in disarray. Myocardial scarring and growth of the collagen matrix also occur.1 The mechanism of scarring continues to be elucidated but appears to reflect small intramural coronary arteriole dysplasia.9
SYMPTOMS AND CLINICAL COURSE
While the most common symptom of HCM is dyspnea on exertion, the majority of patients with HCM are asymptomatic.10 Importantly, symptoms are not always concordant with severity of LV outflow tract obstruction and may be more closely related to diastolic dysfunction.4
With diastolic dysfunction, the increased chamber thickness in HCM results in increased left ventricular (LV) stiffness, impaired filling, and relaxation. These diastolic abnormalities result in elevated left atrial, LV end-diastolic, and pulmonary pressures. Symptoms may also be caused by mitral regurgitation from SAM of the mitral valve, LVOT obstruction, arrhythmias such as atrial fibrillation, and myocardial ischemia. Patients may also complain of chest pain with exertion, syncope or near syncope, or palpitations. Eating may make symptoms worse because of splanchnic vasodilation and the resulting decrease in systemic vascular resistance.11
The clinical course of HCM is variable. In one community cohort, 23% of subjects with HCM had normal life expectancy.12 Other patients may have premature death. Annual mortality rate from HCM is approximately 1%.1 Congestive heart failure and atrial fibrillation may be part of the natural history of HCM. SCD is a frequent and catastrophic initial presentation.13 SCD tends to occur in younger patients and may occur during heavy exertion, light exertion, or even at rest. In an unselected, community-based population with HCM, the estimated incidence of SCD is approximately 0.1% to 0.7% per year.14,15
PHYSICAL EXAMINATION
The physical examination may provide several clues that suggest obstructive HCM. With LVOT obstruction, a harsh systolic murmur exists at the upper sternal border. It is important that this murmur be differentiated from that of mitral regurgitation, which can also be present in HCM secondary to SAM of the mitral valve. Palpation of the carotid pulse aids in distinguishing HCM from aortic stenosis or the presence of a subvalvular aortic membrane. With HCM, little difficulty exists during early systole in ejecting the blood through the LVOT into the aorta; therefore, the carotid upstroke is brisk. As systole progresses, LVOT obstruction occurs, resulting in a collapse in the pulse and then a secondary rise as LV pressure increases to overcome the obstruction. This sign is known as a bisferiens, or spike-and-dome, pulse. In contrast, because the fixed obstruction of aortic stenosis or a subvalvular aortic membrane is present during the entire cardiac cycle, the carotid upstroke in these entities is the classic parvusettardus pulse, a carotid pulse with delayed amplitude and upstroke. Therefore, if any patient carrying a diagnosis of HCM has decreased carotid pulses, this should prompt thoughts of a mistaken diagnosis and further investigation into a fixed obstruction of the LVOT.
Unless congestive heart failure has developed, the lungs are clear and the jugular venous pressure is normal. The point of maximal impulse is often forceful and sustained, and a palpable S4 gallop may be present. Occasionally, a bifid apical impulse may be palpated; the first impulse represents forceful atrial contraction and the second impulse represents sustained ventricular contraction.
The classic auscultatory finding for HCM is a crescendo–decrescendo systolic murmur along the left sternal border that increases with the Valsalva maneuver. The Valsalva maneuver decreases preload, which results in decreased filling of the LV. An underfilled LV results in increased obstruction. The response in HCM to various physiologic and pharmacologic maneuvers is illustrated in Table 22.2.
TABLE
22.2 The Response in HCM to Various Physiologic and Pharmacologic Maneuvers
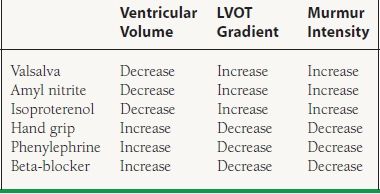
LVOT, left ventricular outflow tract.
During the cardiac examination, it is also imperative to listen carefully for a mitral regurgitation murmur as SAM of the mitral valve frequently causes mitral regurgitation. The remainder of the physical examination is generally unremarkable in HCM.
DIAGNOSTIC TESTING
Labs, Chest X-Ray, and Electrocardiogram
Blood work generally is unremarkable, with the exception of an elevated plasma B-type natriuretic peptide (BNP).16 The chest x-ray is often normal. The electrocardiogram (ECG) may show LV hypertrophy. Occasionally, a pseudoinfarct pattern (with Q waves in the anterolateral leads) may be present on ECG. Figure 22.6 illustrates this pseudoinfarct pattern in a patient with HCM, normal LV systolic function, and no known coronary artery disease. In the apical variant of HCM, the ECG may have deep T-wave inversions in the anteroapical leads (Fig. 22.7). Left atrial abnormality may be present if the patient has had long-standing mitral regurgitation from SAM of the mitral valve. Atrial fibrillation may also be present.
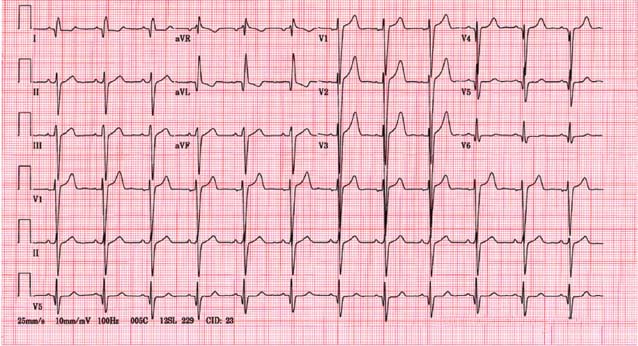
FIGURE 22.6 Pseudoinfarct pattern on ECG in HCM. In HCM, a pseudoinfarct pattern (Q waves in lateral leads) may sometimes be noted. This patient had normal LV systolic function and normal coronary arteries.
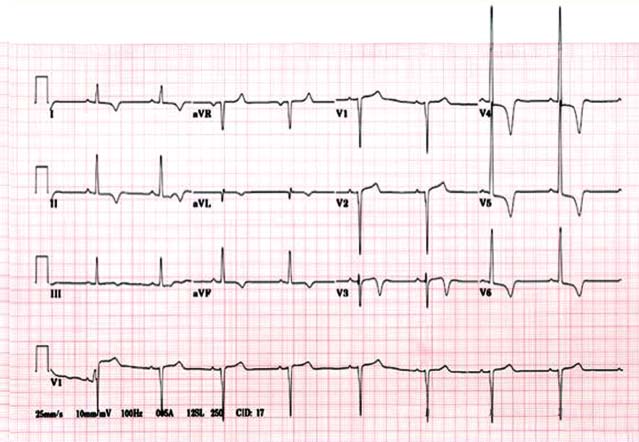
FIGURE 22.7 ECG in apical HCM (Yamaguchi). The classic ECG for apical HCM has deep anteroapical T-wave inversions.
Transthoracic echocardiography (TTE) is currently the primary clinical modality for diagnosing HCM. The septum should be visualized and measured in the parasternal long-axis, apical long-axis, apical four-chamber, and parasternal short-axis views. The major diagnostic criterion for HCM is LV wall thickness of ≥15 mm in the absence of other causes for increased ventricular thickness.4 The LV is nondilated and hyperdynamic. Figures 22.1 and 22.2 are TTE images from HCM patients with marked hypertrophy of the inter-ventricular septum. Figure 22.4 illustrates SAM of the mitral valve and resulting LVOT obstruction. During TTE, particular attention should be paid to the septal thickness; location and pattern of hypertrophy; site and magnitude of LVOT obstruction; presence of SAM of the mitral valve; and presence of premature closure of the aortic valve.
Given the frequency of no obstruction at rest, subjects with suspected HCM should undergo provocative testing during TTE with amyl nitrite, Valsalva, or exercise (treadmill or bicycle) to determine whether latent obstruction exists. Amyl nitrite is a vasodilator that decreases preload to the LV, followed by a compensatory increase in heart rate. Exercise results in an increase in contractility and heart rate. The physiologic effects of amyl nitrite and exercise thus result in an increase in LVOT gradient. In our experience, supervised exercise stress tests in patients with HCM are safe, with a major complication rate of 0.04%.17 Dobutamine is generally not recommended for the purposes of provoking LVOT gradients, for gradients provoked by dobutamine are of questionable clinical significance.4
Pulse-wave Doppler should be performed to record LV and left atrial inflows to assess diastolic function. Diastolic abnormalities, which are common in HCM secondary to the thickness and stiffness of the LV, are unfortunately not specific for the diagnosis of HCM.
The mitral valve should be interrogated in multiple views to assess for the presence of mitral regurgitation, which is commonly present when SAM of the mitral valve leaflet is present (see Fig. 22.4). SAM of the mitral valve has a classic appearance in M mode, where the mitral valve leaflets can be seen to approach and often contact the interventricular septum (Fig. 22.8). In HCM, the mitral valve leaflets may be elongated and anterior displacement of the papillary muscles of the mitral valve may also occur.18 With SAM, the mitral regurgitation may range from mild to severe and is posteriorly and laterally directed in the left atrium because of incomplete leaflet apposition (see Fig. 22.5). If the direction of the color jet of mitral regurgitation is central or anterior, then suspicion should be raised for intrinsic abnormalities of the mitral valve.
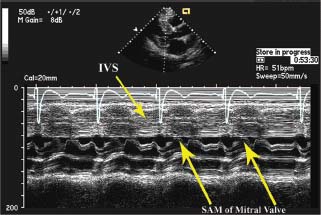
FIGURE 22.8 M-mode of SAM of mitral valve in HCM. With SAM of the mitral valve, the mitral leaflet contacts the interventricular septum during systole in a patient with HCM, as illustrated in this transthoracic M-mode echocardiograph. Normally, the mitral leaflets should be well away from the septum during ventricular systole. IVS, interventricular septum; SAM, systolic anterior motion.
Fixed obstructions such as aortic stenosis, subvalvular aortic membrane, and supravalvular aortic membrane can result in secondary hypertrophy of the interventricular septum, as distinct from the primary hypertrophy of the septum in HCM. In aortic stenosis, the aortic valve is calcified and has restricted mobility, whereas in HCM, the obstruction occurs below the aortic valve, and the aortic valve structure and function are preserved. Subvalvular aortic membranes (Fig. 22.9) and supravalvular aortic membranes may be difficult to visualize on TTE, in which case transesophageal echocardiography may need to be performed to assess for the presence of these structures.
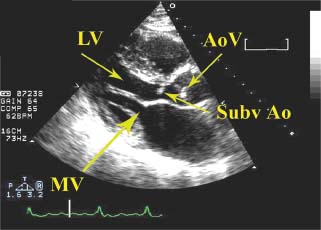
FIGURE 22.9 Subvalvular aortic membrane. Subvalvular aortic membranes must be distinguished from HCM, for both can result in a thickened septum and an LVOT gradient. Although subvalvular aortic membranes may sometimes be difficult to visualize by TTE, in this example a membrane below the aortic valve is clearly seen. LV, left ventricle; MV, mitral valve; AoV, aortic valve; SubvAo, subvalvular aortic membrane.
Continuous-wave Doppler imaging aids in the differentiation of HCM from fixed obstructions. The modified Bernoulli equation [pressure = 4 x (velocity)2] is used with the continuous-wave Doppler tracing through the LVOT to calculate the LVOT gradient. Figure 22.10 illustrates the difference between continuous-wave Doppler signals from HCM and from fixed obstructions. During early systole, blood still flows through the LVOT in HCM; however, with continued contraction of the LV, exacerbated by SAM of the mitral valve, an outflow tract gradient develops. Thus, with HCM, the continuous Doppler signal classically is described as having a late systolic dagger shape, because the obstruction is late peaking as a result of its dynamic nature. In contrast, a fixed obstruction is present during all of systole. Thus, the continuous-wave Doppler signal for fixed obstructions is a smoother contour that peaks earlier.
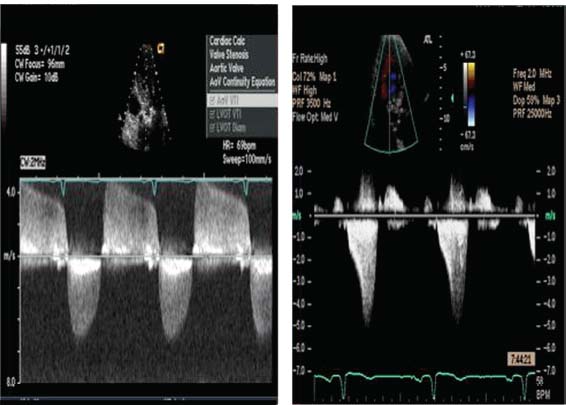
FIGURE 22.10 Continuous-wave Doppler profile comparison of aortic stenosis and HCM. Continuous-wave Doppler profiles from TTE for aortic stenosis (left) and HCM (right) are illustrated. The Doppler profile in aortic stenosis has a smooth, symmetric contour because the obstruction is fixed, whereas the Doppler profile in HCM has a late-peaking, dagger-shaped appearance as a result of the dynamic nature of the obstruction, with its peak in mid-late systole.
The continuous-wave Doppler profile of HCM also must be differentiated from that of mitral regurgitation. The mitral regurgitation jet is generally higher velocity (~6 m/s), whereas the LVOT obstruction jet is often in the 4- to 5-m/s range. The mitral regurgitation velocity tracing also has a smoother, symmetric contour, unlike the dagger-shaped profile of HCM. The mitral regurgitation jet may be late peaking because mitral regurgitation may not occur until SAM has occurred, which occurs partway through systole. However, the mitral regurgitation tracing should extend beyond aortic valve closure, up to the point at which mitral forward flow occurs with diastole. In contrast, the LVOT obstruction signal ends at aortic valve closure.
One promising modality is tissue Doppler, which is sensitive for identifying reduced shortening velocities and may help differentiate between HCM and athlete’s heart, as well as between nonobstructive HCM and hypertensive heart disease with LV hypertrophy.19–21 A transmitral E/septal Ea ratio ≥ 15 has been demonstrated to be a predictive indicator for SCD. Another emerging imaging technique is strain imaging using speckle tracking. Strain imaging continues to evolve while offering complementary information on segmental LV function and its relationship with hypertrophy and fibrosis.22,23
Transthoracic Echocardiogram—Distinguishing HCM from Athlete’s Heart
Because preathletic screening is one means by which the diagnosis of HCM is raised, it is imperative to distinguish HCM from athlete’s heart. Several findings on echocardiography help distinguish HCM from athlete’s heart. In HCM, the septal thickness is usually >15 mm, whereas in an athlete’s heart, septal thickness is <15 mm. Left atrial enlargement often occurs with HCM secondary to long-standing mitral regurgitation from SAM of the mitral valve and/or diastolic dysfunction, whereas in an athlete’s heart, the left atrial size should be normal. The LV should not be dilated in end diastole in HCM, whereas in athletes, it is common for LV end diastolic diameter to be >45 mm. Finally, diastolic dysfunction often exists in HCM as a result of the increased ventricular thickness and stiffness, whereas diastolic function should be normal in athletes. If it is still not certain whether a patient has HCM or athlete’s heart, the athlete should stop training; after 3 to 6 months, ventricular hypertrophy will persist with HCM, whereas with athlete’s heart, hypertrophy should regress.
Cardiac magnetic resonance (CMR) has emerged as a highly useful tool in the diagnosis of HCM. It provides a comprehensive evaluation of myocardial anatomy, including those patients with atypical forms of HCM and those with papillary muscle abnormalities.18,24–26 CMR can also assist with identification of alternative diagnoses such as of Fabry disease and cardiac amyloidosis.
CMR can also provide an accurate assessment of LV function.25,27–29 Additionally, the use of gadolinium-based contrast agents can identify the presence and distribution of myocardial fibrosis.30,31 Several recent studies have examined the relationship between scar burden as assessed in CMR and the incidence of SCD, and it remains an evolving area of interest.32,33
Cardiac Catheterization
While the use of cardiac catheterization has become less relevant in the era of echocardiography and CMR, it is still useful as an adjunctive test in cases where there are discordant data from Doppler echocardiography and the physical exam. Cardiac catheterization may reveal concomitant coronary disease prior to septal myectomy and can also delineate the size and extent of the septal perforators prior to alcohol ablation.
Patients with HCM often have no obstructive coronary artery disease. However, they may have thickened vessels and small-vessel disease from increased collagen deposition in the intima and media.1 The mismatch between myocardial oxygen supply and demand, driven primarily by the increased myocardial mass, may then cause myocardial ischemia. Microvascular dysfunction is present in HCM patients and is associated with worse clinical outcomes.34
The left ventriculogram demonstrates cavity obliteration and a hyperdynamic LV. LVOT gradients can be assessed by positioning a JR4 or multipurpose diagnostic catheter near the LV apex and recording ventricular pressures during slow catheter pullback. A pigtail catheter may not give accurate gradient measurements because there are multiple side holes extending along the distal portion of the catheter, in contrast to the JR4 and multipurpose catheters, which provide true end-hole measurements.
HCM physiology is demonstrated after a premature ventricular contraction (PVC) by the Brockenbrough response (Fig. 22.11). In the beat following a PVC, there is increased filling of the LV from the compensatory pause. The augmented preload results in augmented contractility. In patients with HCM, the increased contractility results in subsequent worsening of the LVOT obstruction. Thus, during the beat after the PVC, there is an increase in LV systolic pressure, a decrease in aortic systolic pressure, and thus an increase in the gradient between LV and aorta. In contrast, in normal subjects, the increased contractility associated with the post-PVC beat results in an increase in both LV systolic and aortic systolic pressure, and there is no gradient between the LV and aorta.
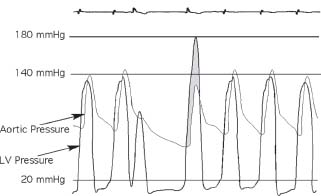
FIGURE 22.11 The Brockenbrough response to a PVC. In normal subjects, a PVC results in a compensatory pause, increased ventricular filling, and subsequent increased cardiac contractility. There is no LV–aortic gradient, either at rest or in the beat post-PVC. The aortic pulse pressure in the beat post-PVC usually increases because of the increased contractility. In contrast, as illustrated in the figure, the Brockenbrough response in the post-PVC beat (fourth beat) suggests HCM. In HCM, the increased contractility occurring with the post-PVC beat results in increased LVOT obstruction and a subsequent increase in the LV-aorta gradient (shaded) as well as decreased aortic pulse pressure during the post-PVC beat.
GENETICS OF HCM
Several hundred mutations involving at least 27 HCM susceptibility genes have thus far been identified.35 The mutations associated with HCM are inherited in an autosomal dominant pattern and primarily involve the myosin, actin, or troponin components of the cardiac sarcomere. The most common mutations that cause HCM involve the β-myosin heavy chain (chromosome 14), myosin-binding protein C (chromosome 11), and cardiac troponin-T (chromosome 1). However, having the HCM genotype does not necessarily imply that subjects will have the phenotypic traits of HCM. Variable penetrance exists, and environmental factors as well as modifier genes affect whether a particular subject will manifest HCM phenotypically.
DNA analysis is the most definitive method for diagnosing HCM.4 With time, genetic testing has become less expensive and readily available.
Traditionally, it has been recommended that first-degree relatives of HCM patients be screened on a 12- to 18-month basis, beginning at age 12 years, with a 12-lead ECG and TTE. The recommended screening interval reflects the fact that latent HCM may be unmasked by growth spurts and subsequent worsening hypertrophy during adolescence. Evidence of late-onset ventricular hypertrophy occurring well into adulthood has spurred a push toward continuing serial echocardiograms past adolescence and into middle age for HCM relatives.36 It is now recommended that adult relatives of HCM patients undergo screening transthoracic echocardiograms at a minimum of every 5 years.
Genetic testing has also been incorporated into the screening process.35 If the patient has a HCM-causing mutation identified, then first-degree relatives can be screened for the presence of that mutation as well. If a relative is mutation positive, then surveillance should be done in a close manner with an annual clinical and echocardiographic exam. If a relative is mutation negative, then casual or no further routine surveillance can be elected provided the TTE is negative and the relative is asymptomatic.
THERAPY
Treatment options for HCM include pharmacologic therapy, septal myectomy, percutaneous alcohol septal ablation, and heart transplantation. Additionally, pacemaker implantation has been performed, but randomized trials have indicated a substantial placebo effect.
Medical Therapy
Treatment with beta-blockers is considered first-line therapy as they improve symptoms and exercise intolerance.37 By decreasing contractile force, beta-blockers decrease the outflow gradient during exercise and decrease oxygen demand. Beta-blockers also lengthen diastolic filling by slowing the heart rate, thus improving any component of myocardial ischemia. We generally start patients on metoprolol, 50 mg twice a day, or extended-release metoprolol, 50 mg daily. If the patient continues to be symptomatic, the dose of metoprolol/extended-release metoprolol can be increased further by 25-mg increments every few weeks. Alternative beta-blocker choices include propranolol, nadolol, or atenolol.
Second-line therapy includes the calcium channel blocker verapamil and the Class IA antiarrhythmic agent disopyramide. Both nondihydropyridine calcium channel blockers and disopyramide exert a negative inotropic effect and improve ventricular relaxation. The extended-release formulation of verapamil can be started at 240 mg daily and increased by 60 mg every few weeks up to 480 mg daily. Calcium channel blockers have been shown to decrease symptoms in comparison to placebo.37 Verapamil should not be used in patients with severe pulmonary hypertension, because this subgroup may develop excessive vasodilation that worsens LVOT obstruction and cardiac output, resulting in pulmonary edema or even death.38 Diltiazem has been used in HCM patients, but there are few data on its effectiveness.
The extended-release formulation of disopyramide may be started at 150 mg twice a day. Disopyramide improves diastolic function and lowers the LVOT gradient.39 Anticho-linergic side effects may occur with disopyramide. Concomitant therapy with beta-blockers is recommended because disopyramide may cause accelerated A–V nodal conduction, which may be deleterious, especially during episodes of atrial fibrillation.
Certain pharmacologic agents should be avoided or used with caution in HCM. Nondihydropyridine calcium channel blockers, such as nifedipine and amlodipine, should be avoided because they cause peripheral vasodilation, which may result in decreased LV filling and worsening of outflow tract obstruction. Angiotensin-converting enzyme inhibitors and angiotensin-receptor blockers, which also cause peripheral vasodilation, should be avoided. Diuretics, if deemed necessary, should be used cautiously, because subjects with HCM often have stiff ventricles that require high filling pressures. Digoxin is not favored in HCM because its positive inotropic effect may worsen LVOT obstruction. Finally, drugs such as dopamine, dobutamine, and norepinephrine can have deleterious effects in the treatment of acute hypotension due to the positive inotropic effects and should not be used. For cases of refractory hypotension that do not respond to IV fluid administration, phenylephrine, a pure alpha agonist that causes vasoconstriction, can be used.
Septal Myectomy
Septal myectomy is considered the most definitive treatment for patients with medically refractory, symptomatic, obstructive (resting or latent gradient of 50 mm Hg or more) HCM.4 In contrast, subjects with gradients >50 mm Hg but no or only mild symptoms are generally treated medically until more severe symptoms manifest. Young patients with marked LVOT obstruction (gradient ≥ 75 mm Hg) should be considered for septal myectomy despite the lack of significant symptoms. In assessing risk and benefit of septal myectomy, the young age of this subgroup decreases the operative risk. Septal myectomy is not indicated in midcavity obstruction. However, in one study, patients with apical hypertrophy complicated by progressive, drug-refractory diastolic heart failure with severely limiting symptoms experienced improved functional status following apical myectomy.40
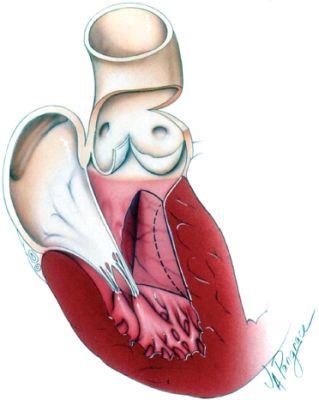
Septal myectomy involves resecting part of the proximal septum through an aortotomy so that the outflow tract obstruction is lessened (Fig. 22.12). Sometimes myectomy may be combined with other cardiac surgery such as coronary artery bypass surgery, mitral valve repair, or mitral valve replacement.
< div class='tao-gold-member'>