Hypertension in the Cardiac Care Unit
Tiberio M. Frisoli
Eyal Herzog
Franz H. Messerli
We have developed at St. Luke’s Roosevelt Hospital Center a novel pathway for the management of arterial hypertension (HTN) in hospitalized patients1 (Figure 33.1).
The Committee on Public Health Priorities to reduce and control HTN in the U.S. Population concluded that “the CDC’s cardiovascular disease program in general, and the HTN program in particular, are dramatically under-funded relative to the preventable burden of disease.” The global burden of HTN is tremendous. HTN is a risk factor for essentially every medical condition that would warrant admission to a hospital’s cardiac intensive care unit (CCU); in fact, the CCU is where the most severe forms of the HTN burden can be seen.
HTN can be defined as a sustained rise in blood pressure (BP) that increases risk for cerebral, cardiac, and renal events. In industrialized countries, the risk of becoming hypertensive (BP >140 per 90 mm Hg) during a lifetime exceeds 90%. The clinical significance of HTN ranges from it being an innocent bystander, to hypertensive emergencies requiring immediate therapy. Patients may be asymptomatic despite marked elevation in systemic BP, yet end-organ consequences of HTN are typically major causes of morbidity and mortality, hence the designation of HTN as the “silent killer.”
HYPERTENSION: DEFINITION AND PATHOPHYSIOLOGY
HTN may be a primary cause for admission to the CCU as in hypertensive emergency. More commonly, however, the CCU patient is admitted for a condition related secondarily to HTN, with high BP as a critical target of therapy and of future risk reduction. For example, while the acute coronary syndrome (ACS) or heart failure (HF) patient is not admitted primarily for HTN, HTN is a mediator of the ongoing myocardial oxygen supply/demand imbalance in ACS, and of pump failure in HF, and thus is a critical target of therapy.
BP is determined by the product of heart rate, stroke volume, and systemic vascular resistance. Heart rate is determined largely by sympathetic activity. Stroke volume depends on cardiac preload and afterload, which in turn depend on multiple variables affecting vascular tone. It follows that BP can be reduced by reducing heart rate, stroke volume, and/or vascular resistance.
HTN can be essential or secondary. Secondary HTN is due to a discernable cause, such as renal or endocrine disease, whereas essential HTN, which is responsible for the most cases, is not. HTN can also be thought of as an elevation of BP caused by one or more abnormalities in cardiac function, vascular function, renal function, and/or neuroendocrine function.
Cardiac: A hyperkinetic circulation caused by excessive sympatho-adrenal activity or increased sensitivity of the heart to baseline neurohormonal regulators increases cardiac output and causes HTN with normal systemic vascular resistance, often in younger patients.
Vascular: Elevated vascular resistance, more common in the elderly, because of abnormal blood vessel responsiveness to sympathetic outflow, endothelial damage which disrupts vasodilatory/vasoconstrictor balance, and ion channel defects will raise BP.
Renal: Renovascular disease leads to increased production of angiotensin II and aldosterone which increase vasomotor tone and sodium/water retention leading to increased cardiac output and systemic vascular resistance.
Neuroendocrine: Neuroendocrine dysfunction as in pheochromocytoma and primary hyperaldosteronism can alter cardiac, vascular, and renal function to cause HTN through a number of various mechanisms.
GENERAL PRINCIPLES ABOUT THE PHARMACOLOGIC MANAGEMENT OF HYPERTENSION
The fundamental concept of HTN treatment is that BP can be decreased by reducing heart rate, stroke volume, and/or systemic vascular resistance. These factors are interrelated, and many pharmacologic agents affect more than one of these determinants of BP. The drugs used to treat HTN are many, but when grouped into classes they are surprisingly few. The armamentarium of drugs used to treat systemic HTN includes those that reduce intravascular volume (diuretics), downregulate sympathetic tone (β-blockers, α-blockers, and central sympatholytics), modulate vascular smooth muscle tone (calcium channel blockers [CCBs] and potassium channel openers), and inhibit the neurohormonal regulators of the circulatory system (angiotensin-converting enzyme [ACE] inhibitors and angiotensin receptor blockers [ARBs]). These drug classes make up the bulk of medications used to treat not only HTN but also the most common disease processes that warrant a CCU admission. β-Blockers, for example, are antihypertensives fundamentally important for reasons that go beyond their antihypertensive effects, in the treatment of HF, ACS, acute neurologic syndrome, and acute aortic syndrome. It is useful to organize drugs in classes and by mechanism of action because an understanding of how the drug class works helps one understand why it is effective for a disease process.
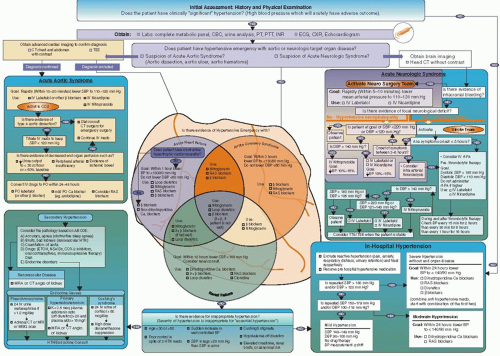 Figure 33.1. Pathway for the management of hypertension for hospitalized patients. |
DIURETICS
Diuretics, which function both to reduce intravascular volume and to vasodilate, increase renal excretion of sodium and water. Thiazide diuretics (e.g., chlorthalidone, indapamide, and hydrochlorothiazide) have long duration of action and moderate intensity of diuresis, suiting them more for chronic HTN treatment than for short-term aggressive diuresis. Once considered firstline for HTN, thiazides are now deemed inferior to several other medications in their antihypertensive potency. Loop diuretics (e.g., furosemide) have a relatively short duration of action (4 to 6 hours) and are useful more for brisk diuresis than antihypertensive efficacy, making them critical in the treatment of HF.
ADRENERGIC BLOCKADE: α– AND β-BLOCKERS
β1 receptors found on myocardial cells, when activated, increase heart rate (chronotropy) and contractility (inotropy). a1 receptors found on vascular smooth muscle promote contraction and thus vasoconstriction; these receptors on the heart increase inotropy.
β-Adrenergic antagonists (e.g., metoprolol) have negative chronotropic and negative inotropic effects as well as effects on resistance vessels. Not only do β-blockers decrease heart rate and BP, but they protect against the cardiac remodeling imposed by adrenergic stimulation. Some drugs (labetalol, carvedilol) block α– and β-receptors thus mediating vasodilatation (α blockade) and decreased reflex tachycardia. By decreasing heart rate and thus increasing diastolic filling time, these drugs increase preload without compromising cardiac output (decreased afterload and increased preload compensate for decreased heart rate).
CALCIUM CHANNEL BLOCKERS
Calcium ions are major mediators of vascular smooth muscle cell contraction and inotropic and chronotropic function of the heart. Calcium enters vascular smooth muscle cells, cardiomyocytes, and pacemaker cells through voltage-dependent calcium channels.2 In vascular smooth muscle, the channel allows entry of sufficient calcium for initiation of contraction by calcium -induced intracellular calcium release from the sarcoplasmic reticulum. In addition to these acute regulatory functions, increased intracellular calcium has atherosclerosis-promoting effects.3
The dihydropyridine CCBs (e.g., amlodipine, nifedipine, felodopine, isradipine) are highly selective for arterial tissues, including the coronary arteries, where they cause vasodilation. Nicardipine has strong antihypertensive activity; intra-arterially it decreases incidence of vasospasm in subarachnoid hemorrhage, and it is a recommended agent for HTN after acute ishemic stroke and intracerebral hemorrhage.4 Clevidipine, a new third-generation intravenous dihydropyridine CCB has a high vascular selectivity with a fast onset and offset of BP lowering effect, thus making it easily and rapidly titratable and an especially attractive drug for acute HTN. In perioperative patients requiring HTN treatment, clevidipine compared favorably to nitroglycerin (NTG), nitroprusside, and nicardipine in terms of BP-reducing efficacy and 30-day outcomes (death, MI, stroke, and renal failure).5
The nondihydropyridine CCBs (diltiazem and verapamil) bind to different sites and are less selective for vascular smooth muscle; they have negative chronotropic and dromotropic effects on sinoatrial and atrioventricular nodal conducting tissue and negative inotropic effects on cardiomyocytes, making them useful heart rate-controlling agents as in atrial fibrillation with rapid ventricular rate.
Nondihydropyridine CCBs should be used with caution in patients with impaired systolic function or conduction system disease because these agents can exacerbate HF and SA or AV node dysfunction, especially in patients already on β-blocker therapy.
ANGIOTENSIN-CONVERTING ENZYME INHIBITORS
ACE inhibitors prevent the ACE-mediated conversion of angiotensin I to angiotensin II, leading to decreased circulating angiotensin II and aldosterone. Angiotensin II elevates BP and promotes target-organ damage, including atherosclerosis, by various mechanisms: direct effects of angiotensin II on constriction and remodeling of resistance vessels, aldosterone synthesis and release, enhancement of sympathetic outflow from the brain, and facilitation of catecholamine release from the adrenals and peripheral sympathetic nerve terminals.6,7
ACE inhibitors decrease levels of vasoconstricting angiotensin II. They also promote natriuresis and reduce intravascular volume. By decreasing bradykinin breakdown, ACE inhibitors promote vasodilatation. They may be nephroprotective because reducing angiotensin II levels reduces renal efferent arteriole constriction thus reducing intraglomerular pressures and limiting glomerular damage over time. For all these reasons, these drugs are particularly useful in the hypertensive diabetic patient and the HF patient.
NITRATES
Organic nitrates are chemically reduced to release nitric oxide (NO), an endogenous signaling molecule that causes vascular smooth muscle relaxation. Although NO can dilate both arteries and veins, venous dilation predominates at therapeutic doses. NO-induced venodilation increases venous capacitance, leading to a decrease in the return of blood to the right side of the heart, and subsequently decreased right ventricular and left ventricular (LV) end-diastolic pressure and volume. This decrease in preload reduces myocardial oxygen demand. At higher concentrations, nitrates may cause arterial vasodilation. In the coronary circulation, NTG preferentially dilates large epicardial arteries rather than smaller coronary arterioles, thus preventing coronary steal phenomenon, encountered with such agents as dipyridamole. It is unclear to what extent the effects of nitrates on coronary vasodilation benefit the patient with angina, because the chronic oxygen deficit in coronary artery disease (CAD) patients causes maximal dilation of coronary arteries, and because atherosclerotic coronary arteries may remain noncompliant even in the face of coronary artery vasodilators. Furthermore, doses of nitrates sufficient to vasodilate epicardial arteries can induce peripheral vasodilation, hypotension, and reflex tachycardia, which harm the delicate supply/demand balance. In patients with HF, however, reflex tachycardia is rare; the venodilation and decreased end-diastolic pressure effects of
nitrates make them acceptable at certain doses for decreasing pulmonary congestion in the patient with hypertensive emergency and congestive HF. Side effects related to hypotension include dizziness and syncope; nitrates are contraindicated in hypotension and not advised in diastolic dysfunction and hypertrophic obstructive cardiomyopathy, two disease states for which adequate preload are critical to sustain cardiac output. Moreover, nitrates should not be taken by patients taking phosphodiesterase inhibitors (i.e., Sildenafil [Viagra]).
nitrates make them acceptable at certain doses for decreasing pulmonary congestion in the patient with hypertensive emergency and congestive HF. Side effects related to hypotension include dizziness and syncope; nitrates are contraindicated in hypotension and not advised in diastolic dysfunction and hypertrophic obstructive cardiomyopathy, two disease states for which adequate preload are critical to sustain cardiac output. Moreover, nitrates should not be taken by patients taking phosphodiesterase inhibitors (i.e., Sildenafil [Viagra]).
Sodium nitroprusside is a nitrate that, such as NTG, liberates NO but does so nonenzymatically; as a result nitroprusside does not target specific vessels and consequently dilates both arteries and veins. With rapid onset of action and high efficacy, nitroprusside is useful in hypertensive emergencies but must be infused with continuous BP monitoring. Sodium nitroprusside is metabolized to products which are potentially toxic if they accumulate excessively: cyanide (acid-base disturbance and cardiac arrythmia) is converted to thiocyanate (psychosis, spasms, and convulsions).
HYPERTENSION AND THE CCU
HTN is a risk factor and target of therapy (both short- and longterm) for virtually every condition that warrants a CCU admission. We will review the most common CCU disease states in terms of pathophysiology, diagnosis, and management.
HYPERTENSION AND DISEASES OF THE AORTA
Diseases of the aorta include aortic atheroma, aneurysm, arteritis, and acute aortic syndromes (dissection, intramural hematoma, and penetrating ulcer). These diseases tend to follow an indolent asymptomatic progressive course until they culminate in an acute life-threatening clinical presentation. The most common of these processes is aortic dissection.
Aortic dissection is characterized by an intimal tear and formation of a false lumen within the media of the artery. Repetitive hemodynamic forces produced by the blood ejected into the aorta with each cardiac cycle contribute to the weakening of the aortic intima and to medial degeneration. Sustained HTN intensifies these forces. Acute elevations in heart rate and BP can lead to catastrophic acute worsening of the disease process. The false lumen, as it extends, can obstruct the true lumen leading to such complications as acute myocardial infarction, acute renal failure, and stroke. Many patients with aortic dissection have undermining of the elastic or muscular components of the media which is quantitatively and qualitatively more severe than expected from aging, as seen in such disease processes as Marfan and Ehler-Danlos syndromes, and congenital bicuspid aortic valve.
Patients with aortic syndrome present with acute onset of severe chest pain which typically radiates through the back. The pain is at its most severe at onset, unlike the typically crescendo pain of ACS. Although clearly not diagnostic, terms such as “tearing,” “ripping,” and “stabbed me in the chest and back” should alert the caregiver to the possibility of dissection. Presentations can vary and thus aortic disease must be considered in any patient presenting with chest pain, especially because some treatments for ACS (thrombolysis) can worsen acute aortic syndrome.
The modalities available for the diagnosis of aortic dissection include CT, MRI, transthoracic echocardiogram (TTE) or transesophageal echocardiogram (TEE), and aortography. The goals of imaging are to establish the diagnosis, determine location of the dissection (ascending, type A dissections are considered surgical emergencies), and determine extent of disease (e.g., coronary or cerebral artery involvement). The choice of modality may be influenced by clinical presentation. The patient with suspected aortic dissection who also presents with physical exam findings suspicious for pericardial effusion or aortic regurgitation may benefit more from a TEE than CT, for example.
Therapy for aortic dissection aims to halt progression; lethal complications arise not because of the tear itself but to extension of the tear such that occlusion or rupture occur. Untreated, aortic dissection will result in death in 25% of patients in 24 hours, 50% in 1 week, and more than 90% in 1 year. As the major etiologic component of the development and progression of aortic dissection, BP is the major target of treatment. Heart rate is also a major target of treatment because BP and heart rate together create hemodynamic force which favors extension of the dissection.
PATHWAY MANAGEMENT
When the dissection involves the ascending aorta, the focus is on surgical management (Figure 33.2). However, whether the dissection is ascending or descending, medical management to reduce BP and heart rate are critical and urgent. Labetalol is an ideal drug as it reduces BP and heart rate, and can be given intrave-nously for immediate onset of action and titration. We recommend lowering BP within 10 to 20 minutes to a systolic BP level of 110 to 120 mm Hg. Esmolol, with its short half-life, may be a good alternative in those with possible intolerance to β-blockers (reactive airway disease). The CCB nicardipine is an alternative as well. Once the patient is stabilized, whether surgically for ascending or medically for descending dissection, oral forms of the same medications (β-blocker, CCB, and ACE inhibitor) are used for longterm BP control, which is crucial for secondary risk reduction.
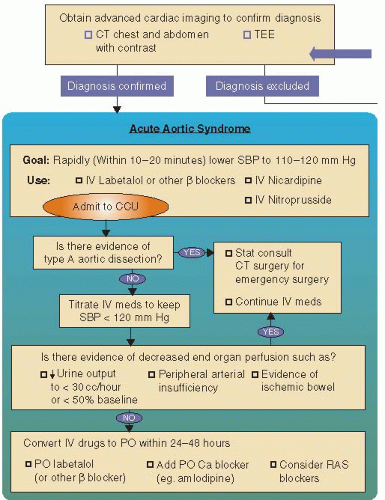 Figure 33.2. Management of hypertension in acute aortic syndrome. |
HYPERTENSION AND ACUTE NEUROLOGIC SYNDROME
Stroke results from either ischemic or hemorrhagic causes. Ischemic stroke can be categorized by presumed mechanism of ischemia into coagulopathies (protein C deficiency, antiphospholipid antibody), small vessel-lacunar (vasculitis, embolic), large vessel-intracranial (dissection, atherosclerosis), large vessel-extracranial (Takayasu, atherosclerotic), and cardioembolic (atrial fibrillation, ventricular thrombus, infectious endocarditis). Some strokes are cryptogenic, or of undetermined cause. Hemorrhagic stroke can be categorized as intracerebral (bleeding into brain parenchyma, further classified as primary and unrelated to a brain lesion, or secondary and related to a congenital or acquired brain lesion or abnormality), subarachnoid, or intraventricular. HTN is a major risk factor for both ischemic (especially small vessel-lacunar) and hemorrhagic stroke, and thus there is little doubt that treating BP for stroke prevention (both primary and secondary prevention) is justified and very important. However, it is in the treatment of HTN for the patient in the acute phase of a stroke that there has been much controversy and ambiguity; recent data is shedding more light on this important issue.
Stroke is a major cause of morbidity and mortality; it is the third leading cause of death in the United States behind heart disease and cancer. Perhaps more devastating is the morbidity in survivors; stroke is the leading cause of long-term disability in adults. Fifteen to thirty percentage of stroke survivors are permanently disabled, and 20% require institutional care for 3 months.
Timely diagnosis of stroke, greatly dependent on history and physical exam, is critical because early implementation of treatment can improve prognosis; some treatments for ischemic stroke, for example, are only implementable within 3 (intravenous recombinant tissue plasminogen activator rtPA) to 6 hours (intra-arterial thrombolysis) from onset of event. For this reason, the most important piece of history is time of symptom onset. Findings such as focal neurologic deficit, persistent neurologic deficit, acute onset of symptoms, and no history of head trauma favor the diagnosis of stroke; the presence of all four of these findings makes stroke about 80% likely. Symptoms also enable the experienced clinician to determine the vascular distribution (e.g., anterior or posterior) of a stroke. Once stroke is suspected, severity can be assessed with the NIH Stroke Scale, the diagnosis can be further pursued with imaging studies, and tests can be obtained to exclude stroke mimics (tumor, hypertensive encephalopathy, seizure, and migraine variant).
The first imaging test to obtain in the acute stroke patient is a noncontrast CT scan of the brain, because this will identify most intracranial hemorrhage and helps exclude stroke mimickers such as neoplasm. Importantly, CT will not reliably diagnose ischemic stroke when early in its evolution; MRI, on the other hand, will. MRI with diffusion-weighted and perfusion-weighted imaging has a high sensitivity and specificity for ischemic lesions, even within minutes of symptoms onset, and can identify brain tissue that is ischemic but not yet infarcted and therefore amenable to being rescued if successfully revascularized. CT angiogram provides maps of cerebral blood flow thus possibly revealing occlusion or stenosis.
An elevated BP is often detected in the first hours after stroke. For every 10 mm Hg increase above 180 mm Hg, the risk of neurologic deterioration increases by 40% and the risk of poor outcome increases by 23%. Despite this association between elevated BP and poor outcomes, the management of arterial HTN in ischemic stroke has been controversial. Investigators found a U-shaped relationship between death and admission BP; both elevated and low admission levels were associated with high rates of early and late death from stroke.8 Elevations in mean BP during the first days after stroke had an unfavorable effect on outcomes; death caused by brain injury and brain edema correlated with high initial BP. Theoretical reasons for lowering BP include reducing brain edema, reducing risk of hemorrhagic transformation, and preventing further vascular damage. Conversely, it has been theorized that overly aggressive BP reduction may lead to neurologic worsening.9 Furthermore, most of the patients experience a drop in BP within the first hour after stroke without any medical treatment. It has thus been suggested that unless thrombolysis is utilized or there is evidence of other BP-related organ dysfunction, there is little scientific evidence and no clinically established benefit for rapid lowering of BP among persons with acute ischemic stroke.10 Thus, in 2007, an expert panel recommended that antihypertensives should be withheld for acute ischemic stroke, unless BP is more than 220/120 mm Hg11; the panel acknowledged that their recommendation was unfortunately not based on very strong or convincing data.
Recent data suggests this “permissive hypertension” may be a mistake. In a study of more than 2,000 ischemic strokes in Mongolia, a systolic BP >200 mm Hg on average 9.3 hours after event onset had an odds ratio of 4.36 for adjusted mortality.12




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
