Gene
Protein function
Phenotype
Status
References
Transcription factors and cofactors
BCOR
Transcriptional corepressor
OFCD syndrome (with DORV)
2 or more independent reports
FOXC1
Forkhead TF
TOF
2 or more patients
[62]
FOXC2
Forkhead TF
TOF
Single case report
[62]
FOXH1
Forkhead TF
TOF
2 or more patients
[44]
GATA4
GATA binding TF
TOF
2 or more independent reports
GATA6
GATA binding TF
TOF
2 or more independent reports
HAND2
Helix-loop-helix TF
TOF
2 or more independent reports
HOXA1
Homeobox TF
BSAS (with TOF), ABDS (with TOF)
2 or more independent reports
NKX2-5
Homeobox TF
TOF
2 or more independent reports
DORV, heterotaxy (with DORV)
2 or more independent reports
NKX2-6
Homeobox TF
TOF
Single case report
[75]
DORV
Single case report
[75]
PITX2
Homeobox TF
DORV
Single case report
[76]
SALL4
Zinc finger TF
Okihiro syndrome (with TOF)
Single case report
[77]
TBX1
T-box TF
TOF, DiGeorge syndrome (with TOF)
2 or more independent reports
TBX5
T-box TF
Holt-Oram syndrome (with TOF)
2 or more independent reports
Holt-Oram syndrome (with DORV)
Single case report
[80]
TBX20
T-box TF
TOF
Single case report
[81]
TFAP2B
AP-2 TF
TOF
Single case report
[82]
ZFMP2
Zink finger TF
TOF
2 or more independent reports
DORV
2 or more independent reports
ZIC3
Zink finger TF
DORV, heterotaxy (with DORV)
2 or more independent reports
Genes involved in signaling pathways
ACVR2B
Activin receptor
Heterotaxy (with DORV)
Single case report
[86]
ALDH1A2
Aldehyde dehydrogenase
TOF
2 or more patients
[87]
BRAF
Serine/threonine protein kinase
LEOPARD syndrome (with TOF)
Single case report
[88]
CFC1
Ligand (EGF family)
TOF
2 or more patients
[44]
DORV
Single case report
[89]
GDF1
Ligand (BMP/TGF-beta family)
TOF
2 or more patients
[90]
DORV
Single case report
[90]
JAG1
Notch ligand
TOF, Alagille syndrome (with TOF)
2 or more independent reports
MAP2K1
MAP kinase kinase
CFC syndrome (with TOF)
Single case report
[92]
NODAL
Ligand (TGF-beta family)
TOF
2 or more patients
[40]
DORV
2 or more independent reports
NOTCH2
Notch receptor
Alagille syndrome (with TOF)
Single case report
[94]
RAF1
Serine/threonine protein kinase
Noonan syndrome (with TOF)
Single case report
[52]
TDGF1
Co-receptor (TGF-beta signaling)
TOF
2 or more patients
[44]
Other genes
CHD7
DNA helicase
CHARGE syndrome (with TOF)
2 or more patients
[55]
CHARGE syndrome (with DORV)
2 or more patients
[55]
GJA5
Gap junction protein
TOF
2 or more patients
[31]
NPHP4
Ciliary protein
DORV, heterotaxy (with DORV)
2 or more patients
[45]
SH3PXD2B
Adapter protein
FTHS (incl. DORV)
2 or more patients
[95]
32.4.1 Cardiac Transcription Factors
Cardiac development is a finely tuned process regulated by transcriptional networks that are governed by a core set of cardiac transcription factors (see Chap. 11), many of which are associated with isolated CHD [6]. The gene NKX2-5 encoding the transcription factor NK2 homeobox 5 is found mutated in about 4 % of TOF patients [33] and also rarely in DORV patients. Mutations in GATA4, which interacts with NKX2-5 at cardiac promoters, primarily cause septal defects [6, 33] but have also been identified in several cohorts of sporadic TOF patients as well as one study of familial TOF [34]. Moreover, mutations in TOF patients have also been reported for the GATA6 gene [35] and HAND2 encoding heart and neural crest derivatives expressed 2 protein [36], a downstream target of GATA4. The activity of GATA proteins is modulated by the FOG family of transcription factors and mutations in the zinc finger protein FOG family member 2 (ZFPM2/FOG2) have been found in various studies on TOF and DORV patients [37, 38]. Furthermore, T-box transcription factors also play an important role in cardiac developmental processes. Haploinsufficiency of TBX1 is the primary cause of CHD in DiGeorge syndrome, and mutations in this gene have also been identified in patients without the 22q11 deletion [12, 19]. Furthermore, mutations in TBX5 cause Holt-Oram syndrome, for which 20 cases with TOF have been described so far [39] and which can also present with DORV.
32.4.2 Genes Associated with Laterality Determination
The bilateral symmetry of the developing embryo is first broken during cardiac development, and left-right patterning is established during early embryogenesis by the interplay of different signaling pathways and by the action of nodal cilia [6] (see Chap. 7). Components of the Nodal signaling pathway are critical in determining organ laterality, and disruption of these genes causes a wide variety of cardiac malformations, often associated with laterality defects [6] (see Chap. 38). Many DORV patients show laterality defects such as heterotaxy, and a number of mutations have been identified in the Nodal pathway [3]. Nodal growth differentiation factor (NODAL) mutations were reported in two studies on DORV patients and could also be identified in TOF patients [40]. The CFC1 gene, encoding the CRYPTIC protein (a co-receptor in Nodal signaling), was found to be mutated in isolated TOF and DORV cases. Moreover, non-synonymous variants that could act as susceptibility alleles were identified in patients with DORV and heterotaxy [41, 42] (see Chap. 38). Further genes encoding signaling molecules of the Nodal pathway that have been found mutated in DORV and/or TOF patients are growth differentiation factor 1 (GDF1), teratocarcinoma-derived growth factor 1 (TDGF1), and activin A receptor, type IIB (ACVR2B). Furthermore, mutations in the transcription factors Zic family member 3 (ZIC3; acting upstream of Nodal signaling) and forkhead box H1 (FOXH1; a possible link between the BMP and Nodal pathways) could be found in several DORV and TOF patients, respectively [43, 44]. Finally, the cilia-related gene NPHP4 encoding nephronophthisis 4 protein was identified in a linkage analysis of a consanguineous family with laterality defects and DORV and was subsequently found mutated in further DORV patients [45] (see Chap. 38).
32.4.3 Genes Involved in Signaling Pathways
Several signaling pathways are involved in the coordination of cardiac development, and mutations of signaling molecules have been identified in isolated and syndromic forms of CHD [6, 8]. Notch signaling is required for patterning of the cardiac chambers and valves and moreover regulates cardiomyocyte proliferation and differentiation [46]. Mutations in the Notch ligand gene JAG1 cause the multisystem disorder Alagille syndrome (AGS), which includes CHD in about 90 % of patients [6]. Approximately 10 % of AGS patients present with TOF [47], and moreover, JAG1 mutations have also been identified in isolated TOF cases [48] and in one study on familial TOF [49]. Interestingly, a JAG1 mutation also was found in a patient with DiGeorge syndrome and TOF, indicating that it might act as a modifier of the phenotype [50]. About 1 % of AGS cases are caused by mutations in NOTCH2 [6], which has been shown for one TOF patient so far. Mutations in the rat sarcoma viral oncogene homolog/mitogen-activated protein kinase (Ras/MAPK) pathway, which regulates cell proliferation, differentiation, and survival, cause Noonan syndrome (NS) and other distinct but overlapping syndromes (see Chap. 23). NS presents with TOF in about 4 % of patients [8, 51], and a microduplication encompassing v-raf-1 murine leukemia viral oncogene homolog 1 (RAF1) has been identified in a patient with NS phenotype including TOF [52]. Moreover, mitogen-activated protein kinase kinase 1 (MAP2K1) and v-raf murine sarcoma viral oncogene homolog B (BRAF) mutations were reported in one TOF patient with cardiofaciocutaneous syndrome and one with LEOPARD syndrome, respectively. Finally, retinoic acid (RA) signaling is both a regulator and target of TBX1, thus playing a role in pharyngeal arch development [6]. Mutations in ALDH1A2, encoding an aldehyde dehydrogenase involved in RA signaling, were identified in two non-syndromic TOF patients.
32.4.4 Epigenetic Regulators
Epigenetic mechanisms represent an important layer of transcriptional regulation and play a central role in cardiac development [53]. Mutations in the CHD7 gene, encoding chromodomain helicase DNA binding protein 7 whose binding correlates with H3K4 methylation, cause CHARGE syndrome. Cardiac malformations occur in 75–80 % of the patients, with TOF being the most common defect. Moreover, DORV with atrioventricular canal is also frequently seen in CHARGE patients [54, 55]. Furthermore, an enrichment of de novo mutations in histone-modifying enzymes could recently be demonstrated in a large CHD cohort including TOF and DORV patients and their parents [56].
32.5 Oligogenic Defects
The majority of TOF cases are isolated, non-syndromic cases – the precise causes of which are yet to be discovered. This is true for the majority of CHDs and many serious non-Mendelian diseases with a clear genetic component. It has been assumed that CHDs might also be caused by rare autosomal recessive variations in concert with private variations [8, 96], which might individually show minor functional impairment but in combination could be disease causing [97]. In this concept, multiple mutations in different genes can lead to disturbances of a molecular network that result in a common phenotypic expression. TOF was the first CHD for which this concept was discovered to be correct [5]. Rare and private mutations in neural crest, apoptotic, and sarcomeric genes were shown to define the genetic background of isolated, non-syndromic TOF (see Fig. 32.1). A great challenge, however, is the discrimination of causative genes. Recently, this challenge was successfully addressed using a burden analysis approach named GMF (gene mutation frequency), which considers the frequency at which a gene is affected by selected deleterious variations in a cohort [5]. Interestingly, besides known developmental genes, cardiomyopathy genes have been confirmed to form part of the genetic basis of TOF. This discovery harbors the possibility that the genetic background of TOF might be correlated to its clinical long-term outcome. Large-scale sequencing projects are currently ongoing and their outcome is of utmost interest.
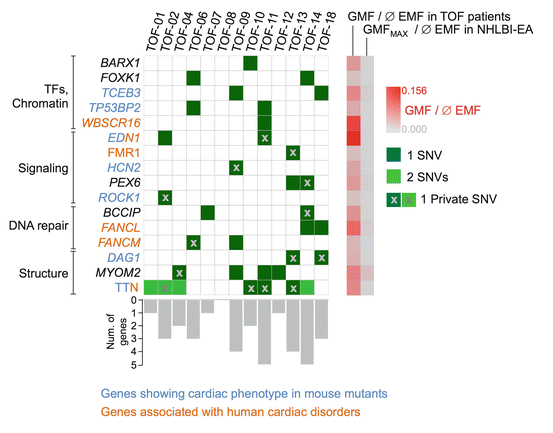
Fig. 32.1
Distribution of mutations found in 16 significantly affected genes (P > 0.05) in TOF patients. Private mutations are marked by ‘x’. Gene-wise frequencies of mutations are represented by gray bars. Gene mutation frequency (GMF) in TOF cases and European-American controls (NHLBI-EA) are indicated by a gray-to-red gradient. For titin (TTN), the average exon-mutation frequency (EMF) over all significantly over-mutated exons is given. EMF exon mutation frequency, GMF gene mutation frequency, SNV single nucleotide variation (adapted from Grunert et al. [5])
32.6 Associations with Common Variations
Besides rare damaging mutations in cardiac regulators and signaling pathways, common SNPs have also been found associated with CHD in several genome-wide association studies (GWAS). Two associated loci (12q24 and 13q32) could be identified in a two-stage study of more than 1600 TOF patients, demonstrating that common genetic variation influences the risk of TOF [57]. However, the majority of CHD-associated variants are individually unique, which results in allelic heterogeneity and reduces the power of GWAS for CHD [58].
Conclusion
Huge advances have been made in understanding the etiology of congenital heart malformations. However, the underlying causes for the majority of CHDs still remain unclear. Estimates suggest that 80 % of cardiac malformations are caused by the interaction of various genetic, epigenetic, and environmental factors [59], which complicates studies aiming to identify single contributors. About 25 % of TOF patients have chromosomal abnormalities such as trisomy 21 or 22q11 deletion, while the majority are isolated cases caused by mutations in cardiac transcription factors, signaling pathways, or most likely combinations of different genes. For DORV, the proportion of patients with chromosomal abnormalities is higher. Moreover, the malformation often occurs in combination with laterality defects like heterotaxy, which is also reflected by the high number of DORV-associated genes playing a role in laterality determination. Taken together, further studies will be required for a deeper understanding of CHD etiology, which will hopefully offer novel preventive and therapeutic strategies and help to improve genetic counseling for affected families.
Acknowledgments
This work was supported by the European Community’s Seventh Framework Programme contract (“CardioNeT”) grant 289600 to S.R.S and the German Research Foundation (Heisenberg professorship and grant 574157 to S.R.S.). This work was also supported by the Berlin Institute of Health (BIH-CRG2-ConDi to S.R.S.).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


