Phenotype
Gene
Associated syndrome
Extracardiac features
Causative gene
Calcific aortic stenosis
NOTCH1
no
no
no
Supravalvar aortic stenosis
ELN
Williams–Beuren
Elfin facies, mental deficiency, overfriendly personality, short stature, renal artery stenosis
ELN
Bicuspid aortic valve
NOTCH1 NKX2-5
SMAD6
Williams–Beuren
Alagille
Kabuki
As above
Jaundice in early infancy, characteristic facies, butterfly vertebrae, ocular anomalies
Distinctive facies, mental retardation, skeletal abnormalities, recurrent otitis media in infancy
ELN
JAG1
MLL2
Aortic coarctation
NKX2-5
SMAD6
Turner
Kabuki
Ovarian failure, short stature, dysmorphic stigmata, neck webbing
As above
MLL2
Interrupted aortic arch
–
22q11 deletion
CHARGE
Hypocalcemia, immunodeficiency, facial dysmorphia, learning difficulties
Coloboma, heart disease, atresia choanae, retarded growth and development and/or CNS anomalies, genital hypoplasia, and ear anomalies and/or deafness
CHD7
Pulmonary stenosis
GATA4
Noonan
NFNS
Holt–Oram
Renal and hematological abnormalities, developmental delay, short stature, characteristic facies
Overlap of neurofibromatosis and Noonan syndrome features
Abnormalities of upper extremities, thumb anomalies
PTPN11
NF1
TBX5
41.2 Aortic Valve Stenosis
Calcific aortic valve disease traditionally has been regarded as a degenerative process involving passive wear and tear of the valve leaflets. On the contrary, current evidence points to a continuum disease process similar to that of atherosclerosis with active cell proliferation and chronic inflammation resulting in leaflet calcification [2].
Familial predisposition to aortic valve sclerosis has been shown by the Hypertension Genetic Epidemiology Network (HyperGEN) Study Group reporting a sibling recurrence risk ratio of 2.3 with several associated chromosomal regions identified by genome-wide linkage analyses [3]. The above results suggested a polygenic nature of aortic sclerosis with a number of susceptibility genes having potentially differential effects on valve calcification. A different study in Western France focused on the more advanced disease trait of calcified aortic valve stenosis requiring surgical replacement to identify clusters of large families segregating aortic stenosis [4].
Congenital malformation of the aortic valve is an additional major risk factor for aortic stenosis [5]. In 2005, Garg et al. performed a genome-wide association study in two kindreds affected by congenital heart disease (CHD) and aortic valve calcification [6]. The first pedigree was a five-generation European-American family in which nine affected members had aortic valve disease (six had bicuspid aortic valve and seven had calcific aortic stenosis). Other cardiac malformations included ventricular septal defect, tetralogy of Fallot, and mitral stenosis. A genome-wide scan in all available family members revealed linkage to a single locus on chromosome 9q34-35 including NOTCH1 gene with a nonsense mutation in affected individuals. Additional screening of a smaller kindred of Hispanic origin revealed a frameshift NOTCH1 mutation in three affected members with bicuspid aortic valve (BAV) [6]. NOTCH1 gene encodes a transmembrane protein regulating cell fate decisions involved in the development of the atrioventricular canal, ventricular myocardium, and cardiac outflow tract [7]. NOTCH1 signaling pathway also represses Runx2 (runt-related transcription factor 2), a critical transcription factor involved in osteoblast differentiation, which is upregulated in human calcified aortic valves [2]. Thus, mutations in NOTCH1 may have a key role in aortic stenosis, resulting in an early developmental defect and a later derepression of calcium deposition in the aortic valve.
Following these findings, Mohamed et al. investigated the role of NOTCH1 gene mutations in 48 German patients with sporadic BAV [8]. A mutation analysis was performed on all 34 coding exons and adjacent intronic and 5′ and 3′ untranslated sequences. Two BAV-causing mutations were identified (p.Thr596Met and p.Pro1797His); both patients had calcification of the aortic valve and concomitant aneurysms of the ascending aorta. These mutations could not be identified in 327 healthy controls.
41.3 Supravalvar Aortic Stenosis
Congenital supravalvar aortic stenosis (SVAS) belongs to a different disease spectrum of diffuse arteriopathy characterized by medial and/or intimal thickening of the aortic wall with narrowing of its lumen [9]. SVAS is commonly a feature of Williams–Beuren syndrome (WBS) with an estimated prevalence of approximately 70 % but can also occur in nonsyndromic forms [10]. Both WBS and nonsyndromic SVAS are caused by disruptions in the elastin (ELN) gene with reduced ELN expression found in vascular smooth muscle cells (VSMCs) of affected patients [11]. Reductions in elastin, a key structural protein of the extracellular matrix, lead to elastic fragmentation with increased collagen and VSMC hypertrophy in the media of SVAS patients [9].
41.4 Aortic Coarctation and Bicuspid Aortic Valve
Aortic coarctation accounts for 5–7 % of CHD lesions [12, 13]. More than 50 % of patients with coarctation of the aorta are also found to have a BAV [12, 13] (Fig. 41.1). The latter is the commonest congenital heart lesion affecting 0.5–2 % of the general population and is a more complex trait than initially thought, with coexistence of defective valvulogenesis and an additive effect on structural properties of the aortic wall [14, 15].
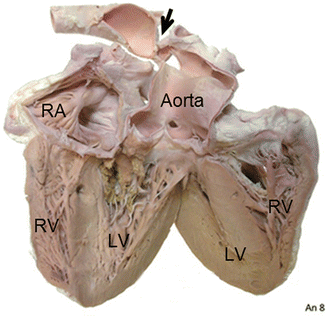
Fig. 41.1
The left ventricle (LV) has been opened longitudinally through the outflow tract and the aortic valve. There is a bicuspid aortic valve with dilatation of the ascending aorta and coarctation (arrow) where the arch narrows discretely. RA right atrium, RV right ventricle (Leon Gerlis Museum, Royal Brompton Hospital, London, UK)
Aortic coarctation and BAV disease can occur in isolation or as part of genetic syndromes, with their underlying genetic basis under ongoing investigation. Both defects frequently can be found in females with Turner syndrome (45X0) characterized by the presence of webbed neck, short stature, and gonadal dysgenesis [16]. Cardiovascular abnormalities are found in up to 45 % of Turner patients, with BAV and aortic coarctation in 30 % and 12 %, respectively [16, 17]. Bileaflet aortic valve can also be a component part of familial thoracic aortic aneurysm or hypoplastic left heart syndrome [18, 19]. Genes located on the short arm of chromosome X are considered key to aortic valve and arch development with happloinsufficiency for Xp associated with higher prevalence of BAV and coarctation in Turner syndrome [17]. A maldevelopment of neural crest cells, which give rise to the outflow tract of the heart, the aortic arch system, and the cervicocephalic arteries, may also underlie development of these lesions in nonsyndromic cases [20].
The presence of BAV can comprise a feature of a number of additional syndromes, such as Alagille, Williams–Beuren, and Kabuki syndromes. Alagille syndrome (AGS) consists of jaundice in early infancy, characteristic facies, butterfly vertebrae, ocular anomalies, and a wide spectrum of cardiovascular anomalies, the most common of which being stenosis or hypoplasia of the branch pulmonary arteries. Mutations in the jagged 1 (JAG1) gene, encoding a NOTCH receptor ligand, are present in up to 75 % of individuals with AGS and may also be found in patients with only a few characteristics of the syndrome [21, 22]. In 2002, McElhinney et al. reviewed the cardiac phenotype of 200 individuals with AGS or a JAG1 mutation [23]. Two of the probands had BAV disease as a primary cardiovascular anomaly and another two as a secondary anomaly, with JAG1 mutations present in three of the BAV patients. Coarctation of the aorta was also present in four subjects with JAG1 mutations found in two of these. Isolated reports of systemic vascular abnormalities in AGS patients exist in the literature, including intracranial aneurysms as well as coarctation of the thoracic and abdominal aorta, with presence of JAG1 mutations in the latter [24, 25]. Expression of JAG1 has been previously shown to contribute to angiogenesis, both in in vitro models as well as in animal studies, with disruption of the mouse JAG1 gene leading to lethal diffuse hemorrhages [26, 27].
Notably, the presence of a bileaflet aortic valve has also been ‑reported in 11.6 % of patients with a clinical diagnosis of WBS [28]. In 2003, Sugayama et al. used fluorescence in situ hybridization (FISH) methods to detect microdeletions in the elastin gene in 20 WBS patients [29]. All FISH-positive patients (85 %) had cardiovascular malformations, out of which 18 % had combined BAV and SVAS. Another syndromic association of both BAV disease and coarctation of the aorta is that of Kabuki syndrome, characterized by distinctive facies, mental retardation, skeletal abnormalities, and recurrent otitis media in infancy. Congenital heart lesions are present in 31–58 % of affected patients with common occurrence of coarctation of the aorta [30, 31]. A recent study undertaking exome sequencing in a discovery cohort of ten unrelated patients with Kabuki syndrome led to identification of MLL2 (myeloid/lymphoid or mixed-lineage leukemia 2) gene loss-of-function mutations in 90 % of cases, with further targeted sequencing identifying additional mutations in 60 % of a replication cohort [32]. Notably, four out of the ten initially studied subjects had coarctation of the aorta, including three patients with a concomitant BAV. MLL2 (also known as KMT2D) encodes a histone methyltransferase, lysine (K)-specific methyltransferase 2D, regulating the transcription of a diverse group of genes with murine studies of MLL2 loss-of-function phenotype exhibiting increased apoptosis and retarded development [33, 34].
The cardiac homeobox gene NKX2–5 has a major role in cardiac development with mutations in this gene associated with a range of cardiac malformations [35–37]. Targeted sequencing of NKX2–5 in 608 subjects with conotruncal anomalies, left-sided lesions, atrial septal defect, and Ebstein’s anomaly resulted in identification of 12 distinct mutations in 3 % of the studied population, including a single (out of 59) patient with coarctation of the aorta [38]. Moreover, previous murine studies have exhibited that heterozygous NKX2–5 ablation results in an eightfold increase in the prevalence of stenotic BAV [35]. Following these findings, Majumdar et al. performed a targeted mutational analysis of the protein coding sequence of NKX2–5 gene in 19 patients with BAV and ascending aortic aneurysm [39]. DNA was extracted from peripheral blood leukocytes and diseased aortic tissue. Three subjects with normal aortic valves and isolated ascending aortic aneurysm were used as controls. Sequence analysis of leukocyte DNA demonstrated a single known polymorphic alteration in the 3′ untranslated region adjacent to exon 2 in four BAV and two control subjects. The same polymorphism was detected following DNA sequencing from aortic tissue samples, in six BAV and one control subject. Interestingly, in four of the BAV patients (and none of the controls), the mutation was only present in the DNA extracted from the aortic tissue suggesting somatically acquired mutation and clonal expansion during aortogenesis.
Bone morphogenetic protein (BMP) signaling also has a pivotal role in cardiac morphogenesis with mice genetically disrupted for BMP exhibiting hypoplastic cardiac cushion formation [40, 41]. SMAD family member 6 (SMAD6) is an important intracellular inhibitor of the BMP pathway, with its genetic disruption in mice resulting in heart valve defects and aortic ossification [42]. Non-synonymous mutations in SMAD6 gene have been recently found in 3 out of 436 individuals with a wide range of CHD lesions, out of which two had a BAV: a 30-year-old male with aortic stenosis, coarctation, and calcification of the aorta and an infant with moderate aortic stenosis [43]. In a different study undertaking exome sequencing of 362 parent–offspring trios with severe CHD, a significantly higher burden of protein-altering de novo mutations in genes with high heart expression (HHE) was found in CHD patients compared to controls [44]. Out of the above studied population, 10 % had coarctation of the aorta, 11 % had BAV, and 4 % had non-BAV aortic stenosis. One of the most interesting genes highlighted in this study is H3K4me, an epigenetic mark involved in activation of key developmental genes. Mutations in genes involved in the H3K4me pathway accounted for 27 % of pathogenic mutations in the HHE gene set with some examples of the above mutated genes in patients with left ventricular outflow obstructive lesions including MLL2 (frameshift mutation), KDM5A encoding lysine (K)-specific demethylase 5A (missense mutation), and USP44 encoding ubiquitin-specific peptidase 44 (missense mutation).
41.5 Interrupted Aortic Arch
Interrupted aortic arch (IAA) accounts for approximately 0.2–1.4 % of all congenital heart defects and is commonly associated with other cardiac lesions, such as ventricular septal defect, patent arterial duct, BAV, truncus arteriosus, and complete transposition of the great arteries [45, 46]. IAA can occur in the context of coloboma, heart disease, atresia choanae, retarded growth and development and/or CNS anomalies, genital hypoplasia, and ear anomalies and/or deafness (CHARGE) syndrome, with a frequency of 3/299 in a study population carrying pathogenic CHD7 mutations for CHARGE syndrome [47]. However, its commonest association is with 22q11 deletion syndrome with 50 % of IAA patients testing positive for a deletion [48].
The 22q11 deletion syndrome has a wide spectrum of clinical features, including the phenotypes of DiGeorge and velocardiofacial syndrome (DGS/VCFS) [49]. The syndrome is characterized by cardiovascular defects, hypocalcemia, immunodeficiency, facial dysmorphia, and learning difficulties. Common associated cardiac malformations include tetralogy of Fallot, IAA, and truncus arteriosus [50]. Most DGS/VCFS patients are hemizygous for a 1.5–3.0 Mb 22q11.2 deletion encompassing up to 40 genes, including TBX (T-box) 1 with development of conotruncal defects in heterozygous mice for a null TBX1 mutation [51, 52]. Interestingly, the incidence of 22q11 deletions varies significantly between different types of IAA, with approximately half of type B patients (interruption between the left carotid and left subclavian artery) and none of the type A patients (interruption distal to the left subclavian artery) having a deletion, suggestive of differential underlying pathologies [48, 53].
41.6 Pulmonary Stenosis
Congenital pulmonary stenosis can occur in isolation (Fig. 41.2) and in combination with other congenital heart lesions, such as tetralogy of Fallot, or as part of genetic syndromes with cardiac involvement. A characteristic example of the latter is Noonan syndrome, the second commonest syndromic cause of congenital heart disease (CHD) after trisomy 21 [54]. Noonan syndrome is an autosomal dominant trait affecting multiple systems, including renal and hematological abnormalities, developmental delay, short stature, and characteristic facial features. Pulmonary stenosis is found in up to 60 % of affected patients with other associated heart lesions including hypertrophic cardiomyopathy, ventricular and atrial septal defect, aortic and mitral valve lesions, and aortic coarctation [55].
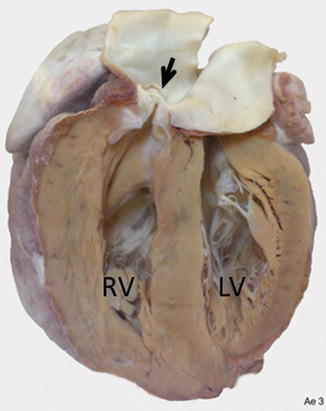
Fig. 41.2




The heart has been cut longitudinally through the right ventricular outflow tract. There is a thickend dome-shaped pulmonary valve (arrow) causing pulmonary stenosis. Flow is through a small orifice. Consequently there is remarkable hypertrophy of the right ventricular wall. RV right ventricle, LV left ventricle (Leon Gerlis Museum, Royal Brompton Hospital, London, UK)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
