Fig. 8.1
Homologous recombination and nonhomologous end joining process
Furthermore, inhibition of RNF8 activity can suppress BRCA1, independent of HR in tumor cells with low 53BP1, and RNF8 could establish a bridge between HR and NHEJ [20]. In RNF8/BRCA1- or RNF8/BRCA1/53BP1-depleted cells, other DNA repair components, including 53BP1, RAP80, and RAD51, cannot assemble at double-strand breaks [20]. Nakada et al. found that the pharmacologic inhibition of RNF8 or RNF168 suppresses HR only in BRCA1-mutated/53BP1-low cancer cells but not in healthy cells and combining DNA-damaging agents and the inhibition of RNF8 may be useful as a cancer therapy [20].
Partner and localizer of BRCA2 (PALB2) is also a breast cancer susceptibility gene and was first identified by its interaction with BRCA2 protein as it is required for the localization of BRCA2 to sites of DNA damage [24] (Fig. 8.1). The BRCA1-PALB2-BRCA2-RAD51 network could be a critical determinant of the responsiveness of specific tumors and individuals to DNA interstrand cross-linking agents. A defect anywhere in this pathway would be expected to result in defective assembly of RAD51 foci, which might be predictive of the responsiveness of a particular tumor to DNA interstrand cross-linking agents [24]. For instance, we can speculate that tumors with low BRCA1 and low 53BP1-RIF1 but high levels of PALB2 can be resistant to platinum-based combinations (Figs. 8.1 and 8.2).
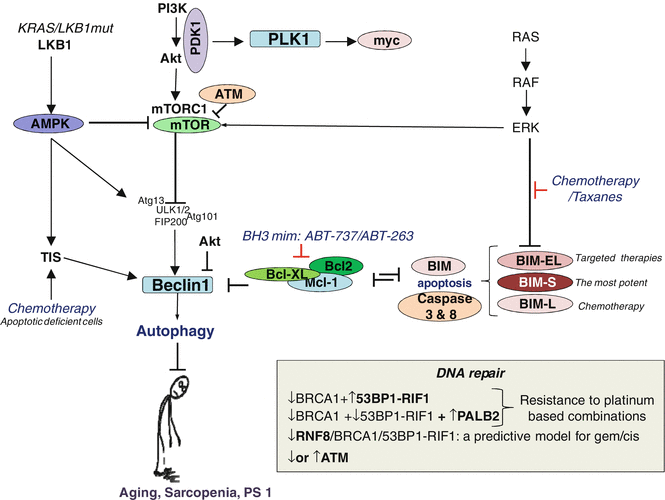
Fig. 8.2
Chemoresistance can occur through cross-regulation between apoptosis, autophagy, and cell senescence
ATM protein lies at the heart of the DDR as a master regulator of three essential DDR processes: cell cycle regulation, DNA repair, and apoptosis [25]. ATM-deficient human tumors frequently display chemotherapy resistance by repairing chemotherapy-induced DSBs through the error-prone NHEJ. NHEJ in these cases serves as a backup mechanism for failed HR-mediated DSB repair in ATM-defective tumors [25]. During NHEJ, the noncatalytic subunits Ku70 and Ku80 form a heterodimer that binds to the free DNA ends and subsequently recruits DNA-dependent protein kinase catalytic subunit (DNA-PKcs). DNA-PKcs kinase activity is essential for the rejoining of the broken ends during NHEJ and can be a valid target for therapy in ATM-defective tumors [25]. Therefore, it would be interesting to test DNA-PKcs inhibitors in patients who have been stratified on the basis of their ATM status.
We are currently examining the mRNA levels of 53BP1, RIF1, RNF8, ATM, and PALB2 as alternative biomarkers that could elucidate DNA repair mechanisms using remaining tissue from patients included in the BREC study.
The Role of Apoptosis, Autophagy, and Cell Senescence
Most chemotherapeutic agents induce apoptosis through two distinct pathways: the intrinsic and the extrinsic apoptotic pathways [26]. The extrinsic apoptotic pathway is triggered by ligand binding to cell-surface receptors. The death-inducing signaling complex promotes activation of caspase 8, which then activates caspase 3. The intrinsic pathway is activated by different apoptotic stimuli that lead to release of cytochrome c from mitochondria and activation of caspase 9 and caspase 3 [26]. The intrinsic mitochondrial apoptosis pathway is controlled by complex interactions between the proapoptotic and antiapoptotic members of the BCL2 protein family. One key proapoptotic BH3-only family member is the Bcl2-interacting mediator of cell death (BIM) [26]. BIM activates cell death either by opposing the antiapoptotic members of the BCL2 family (BCL2, BCLXL, MCL1, and BCL2A1) or by binding to the proapoptotic family members (BAX and BAK1) and directly activating their proapoptotic function [26]. It is well known that several kinase-driven cancers, including chronic myeloid leukemia and EGFR-driven NSCLC, maintain a survival advantage by suppressing BIM transcription and by targeting the BIM protein for proteasomal degradation through MAPK-dependent phosphorylation. Chemotherapy can abrogate such events and induce apoptosis through activation of FOXO3 and its targets, including BIM [26].
Alternative splicing generates at least three BIM isoforms, including BIMS, BIML, and BIMEL, which differ in their proapoptotic activity [27]. Also, a number of studies have raised the possibility that chemotherapy triggers apoptosis by inducing the death-inducing signaling complex [28]. For instance, paclitaxel induces BIMEL accumulation that is required for paclitaxel-induced apoptosis both in vitro and in vivo [29]. Constitutive activation of the MAPK pathway suppresses BIM induction by phosphorylating and inducing proteasomal degradation of BIM, thereby blocking response to paclitaxel [29]. This provides a mechanistic explanation for chemotherapeutic-mediated apoptosis and suggests that combining a proteasome inhibitor with paclitaxel would provide therapeutic benefit among tumors with MAPK pathway activation. A high degree of heterogeneity in BCL-2 family protein levels before treatment may have some value as a predictive marker for cancer response to chemotherapy, but the role of chemotherapy-induced apoptosis, which is a dynamic process, needs to be further explored.
Cell death is most commonly associated with apoptosis, but it can also occur through other mechanisms, including autophagy. A number of studies have reported that autophagy, or autophagic cell death, is activated in cancer cells that are derived from tissues such as breast, colon, prostate, and brain, in response to various anticancer therapies [30]. In the BREC study, a significant interaction between the ECOG Scale of Performance Status (PS) and treatment arm was found in the SLCG BREC study, and a favorable though nonsignificant effect for the experimental arm was observed among patients with PS 0, while patients with PS 1 showed a negative effect for the experimental arm, including a significantly increased risk of death (Moran et al. submitted). PS is widely used to quantify the functional status of cancer patients and is an important factor determining prognosis in a number of malignant conditions. Autophagy is a dynamic process in which intracellular membrane structures sequester proteins and organelles to degrade and turn over these materials. Whether autophagy kills cancer cells or protects them from unfavorable conditions has not yet been clearly answered [30].
Autophagy is the primary cellular catabolic program activated in response to nutrient starvation and amino acid starvation or mTOR inhibition and enhances the initiation of the autophagic flux [31] (Fig. 8.2). Cancer-induced muscle loss and preexisting age-related sarcopenia, both correlated with worse PS, could be a consequence of autophagy impairment. What could the mechanisms by which cancer cells suppress baseline levels of autophagy be? Low expression of Beclin1 or overexpression of mTORC1 or AKT that can inhibit directly Beclin1 can be potential mechanisms of autophagy suppression [31]. Beclin1, a Bcl-2 homology 3 (BH3) domain-only protein, plays an important part in both autophagosome formation and autolysosome fusion. The kinase activity of Beclin1 is negatively regulated by Bcl-2 family proteins, such as Bcl-2 or Mcl-1 Bcl-xL, which bind to Beclin1 and inhibit autophagosome formation [32]. Mammalian Ste20-like kinase 1 (Mst1) is a serine–threonine kinase and a component of the Hippo signaling pathway. Mst1 induces phosphorylation of Beclin1 and promotes binding of Beclin1 with Bcl-2 and/or Bcl-xL. As the interaction between Beclin1 and Bcl-2 and/or Bcl-xL is markedly reduced when Mst1 activity is inhibited, the kinase activity of Mst1 likely regulates the basal interaction between Beclin1 and Bcl-2 and/or Bcl-xL [32]. In addition, ATM activates the TSC2 tumor suppressor via the LKB1/AMPK metabolic pathway in the cytoplasm to repress mTORC1 and induce autophagy [33]. It is well known that autophagy is promoted by AMP-activated protein kinase (AMPK), which is a key energy sensor and regulates cellular metabolism to maintain energy homeostasis and is inhibited by the mammalian target of rapamycin (mTOR), a central cell growth regulator that integrates growth factor and nutrient signals [34] (Fig. 8.2).
Anticancer agents are known to induce cellular senescence which is characterized by an unexpected hypermetabolic phenotype composed of enhanced glycolysis and a discordant negative FLT-PET but positive FDG-PET scan posttreatment [35]. Although therapy-induced senescence (TIS) is a desirable therapeutic outcome, particularly in apoptotically compromised tumors, the viable senescent cells have potentially harmful properties. For instance, they may be capable of cell cycle reentry, affect stemness, secrete paracrine-active factors, and promote inflammation [35]. Therefore, subsequent elimination of senescent tumor cells should add considerably to the long-term efficacy of pro-senescent therapies. The metabolic reprogramming of senescent cells increases protein synthesis and senescence-associated secretable peptides which subsequently increases proteotoxic stress and autophagy. Blocking autophagy in this case can induce apoptosis of senescent cells [35].
Also, very recently, Tan and colleagues elucidated a novel 3-phosphoinositide-dependent protein kinase-1 (PDK1)–Polo-like kinase 1 (PLK1)–MYC signaling pathway connecting two fundamental oncogenic programs, phosphoinositide 3-kinase (PI3K) and MYC. PDK1–PLK1–MYC signaling has a functional role in cancer cell survival and tumor formation. The therapeutic benefit of inhibiting PDK1 and PLK1 pharmacologically in cancer tackles the most undruggable tumors defined by elevated levels of the MYC oncoprotein. PDK1–PLK1–MYC signaling induces an embryonic stem cell-like gene signature associated with aggressive tumor behaviors and resistance to chemotherapy [36] (Fig. 8.2).
We are currently examining the mRNA levels of mTOR, BIM, BIMEL, caspase 8, caspase 3, Beclin1, Bcl-2, Mcl-1, and MYC as biomarkers that can help us to define predictive models for chemotherapy outcome and contribute to synthetic lethality therapeutic approaches.
Conclusions
Platinum-based combination chemotherapy remains the standard first-line therapy for advanced stage NSCLC. The discovery of molecular biomarkers with the potential of selecting patients and predicting drug efficacy is essential in the quest for personalized management of advanced NSCLC. Current results demonstrate that it is difficult to identify a single marker able to predict response to a drug or a combination of drugs. Integrated analysis of several potential biomarkers based on the study of DNA repair pathway biology, but also apoptosis, autophagy, and TIS, will probably provide more insight into predictive modeling in lung cancer.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


