59 High-Risk Vulnerable Plaques
Definition, Diagnosis, and Treatment
Despite a steady decrease in the incidence of acute coronary events, cardiovascular disease (CVD) continues to be the major cause of death worldwide, with 16.7 million deaths per year, representing 29.2% of the total global deaths.1–3 Coronary atherosclerosis is a condition that can be asymptomatic over decades. The transition from asymptomatic, nonobstructive disease to symptomatic, occlusive disease is related to acute coronary thrombosis. This condition, known as atherothrombosis, is mostly related to plaque rupture or erosion, which triggers a series of biochemical events ultimately leading to thrombus formation.4,5 Atherosclerotic plaques at increased risk for rupture have been histologically characterized as thin cap fibroatheromas (TCFA), which can be properly identified by invasive imaging techniques.6 As a result, the concept of early detection and treatment before clinical events has caused increased interest in the catheterization laboratory (cath lab). This chapter, divided into four sections, describes a systematic approach to high-risk vulnerable plaques (VPs). The first section is devoted to definition, incidence, risk factors, and anatomic location of plaque. The second section is devoted to plaque composition and to the pathophysiologic aspects of these lesions, setting up the foundations to understand the dynamics of plaque vulnerability. The third section summarizes the evolving field of invasive plaque imaging, with a brief comment on noninvasive imaging modalities. Finally, the fourth section is devoted to therapy, from conservative, pharmacologic options to aggressive percutaneous coronary intervention (PCI) alternatives.
 Clinical Characteristics
Clinical Characteristics
Definition and Clinical Evidence
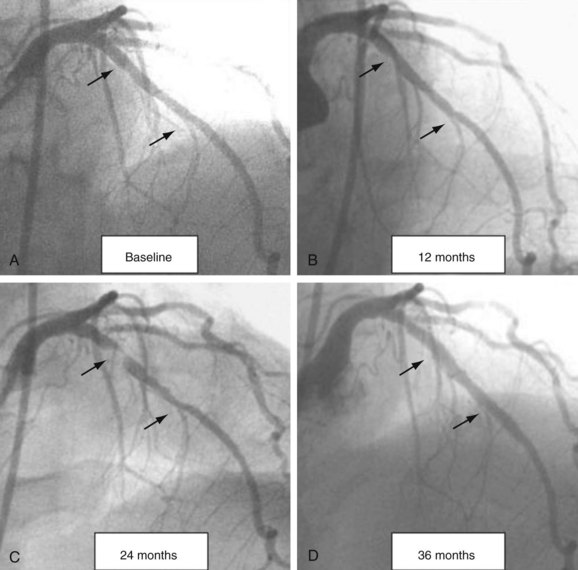
A high-risk VP is a nonobstructive, silent coronary lesion that suddenly becomes obstructive and symptomatic (Fig. 59-1). The initial understanding of this process was described by Ambrose and Fuster in 1988 when they studied the baseline characteristics of nonobstructive lesions that evolved into acute myocardial infarction (AMI).7 The investigators found that at baseline, the lesions evolving to complete occlusion had a mean diameter stenosis of 48%. Multiple investigators reproduced this finding using both angiographic and intravascular imaging modalities and supporting the concept that the majority of lesions responsible for AMI originate from nonobstructive disease.8
Incidence
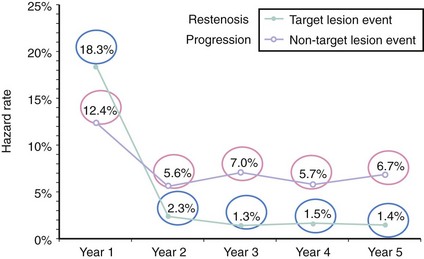
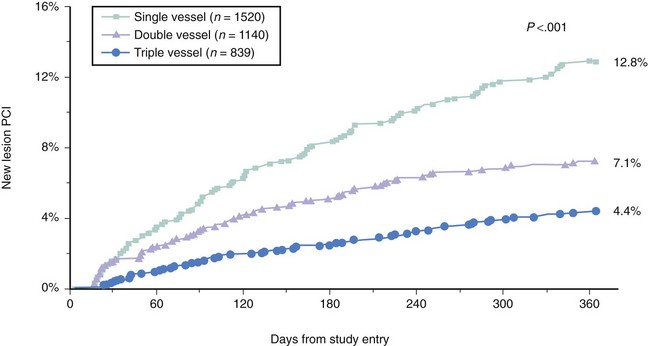
The number of nonobstructive, asymptomatic lesions that progress to obstructive, symptomatic lesions presenting as an acute event can give us the estimated incidence of this condition. Several studies have retrospectively addressed this issue. The first study included 1228 patients who underwent percutaneous coronary intervention (PCI) for symptomatic coronary artery disease (CAD). The incidence of nonobstructive, nonculprit lesions that required additional PCI was 12.4% in the first year and 5% to 7% per year from years 2 to 5 after the initial procedure (Fig. 59-2).9 The second study included 3747 post-PCI patients from the National Heart Lung and Blood Institute (NHLBI) registry.10 The incidence of nonobstructive, nonculprit lesions that required additional PCI was 6% in the first year, ranging from 4.4% to 12.8% according to the number of vessels involved (Fig. 59-3). A third study included a total of 1059 patients studied with computed tomographic angiography (CTA). VPs were classified according to the presence or absence of two variables: (1) positive remodeling, and (2) low attenuation (“soft”) morphology on CT.11 The incidence of acute coronary events was 11% in plaques with one variable and up to 22% in plaques with both variables. Notably, the absence of these two variables represented a strong negative predictive value, with an incidence of coronary events as low as 0.5%. Another study using intravascular modalities recently reported a 12% incidence of TCFAs by optical coherence tomography (OCT) in sites other than the culprit lesion responsible for the acute coronary syndrome (ACS).12 Additionally, they found that the majority of TCFAs occurred in the proximal segments of the coronary vasculature. Despite this incidence of 11% to 22%, the actual area occupied by these plaques in the coronary circulation may not be as large. Cheruvu and Virmani performed detailed histopathology in the entire vasculature of 50 human hearts and demonstrated that only 1.1% to 1.6% of the coronary vessel area was occupied by TCFAs. Out of all the lesions studied, 10.8% corresponded to TCFA.13 This is in agreement with a second study that identified a 11% incidence of TCFAs, with slightly higher incidence in patients with ACS, suggesting a more “unstable” profile of the coronary lesions in these subjects.8,14 These retrospective and pathologic studies gave the foundation to design the first prospective natural-history study of VPs, also called the PROSPECT (Predictors of Response to CRT) Trial.15 A total of 697 patients with ACS underwent three-vessel coronary angiography and gray-scale and radiofrequency (RF) intravascular ultrasound (IVUS) after multi-vessel coronary stenting. The incidence of nonculprit lesions developing major adverse cardiovascular events (MACEs) was 11.6% at 3.4 years. Most of these events were rehospitalization for progressive angina. Cardiac death or MI occurred in only 4.9% of the population. Three independent variables predicted these events and include (1) plaque burden of 70% or greater (hazard ratio [HR] 5.03; P < 0.001); (2) minimal lumen area (MLA) of 4 mm2 or less (HR 3.21; P = 0.001); and (3) RF classification of TCFA (HR 3.35; P < 0.001). Of great interest, plaques exhibiting all three features had a higher HR, up to 11.05 (P < 0.001). Nevertheless, the prevalence of these high-risk plaques was only 4.2%. As a result, we now know that high-risk plaques are uncommon and are composed by a combination of anatomic features, which includes plaque burden and MLA, in addition to TCFA.
Risk Factors
Multiple regression analysis identified multi-vessel CAD at baseline (three- and two-vessel CAD), previous PCI, and age less than 65 years as independent predictors for VP. Of note, treatment with statins failed to protect patients within the first year.10 IVUS studies also identified age, hypertension, diabetes mellitus (DM), heart failure, and dyslipidemia as predictors for IVUS-derived TCFA (ID-TCFA).16 Diabetes is strongly associated with higher rates of ID-TCFA (21.6% vs. 13.1%, P = 0.01), and up to 54.4% in patients with a diagnosis of DM greater than 10 years.17 The incidence of TCFA was also seven times higher in males compared with females.13 In terms of biomarkers and VP, serum myeloperoxidase is a new potential marker of plaque vulnerability.18,19 Similarly, Lipoprotein-associated phospholipase (Lp-PLA2) may play a key role in necrotic core expansion as demonstrated by the Integrated Biomarkers and Imaging Study-2 (IBIS-2).20 In isolated studies, highly sensitive cross-reactive protein (hs-CRP), white blood cell (WBC), interleukin (IL) 18, and tumor necrosis factor alpha (TNFα) inversely correlated with fibrous cap thickness. However, hs-CRP appeared to be the only independent predictor by regression analysis.21 Furthermore, hs-CRP has been postulated as a modulator of neovascularization.22 Nevertheless, the clinical evidence for primary prevention of hs-CRP is rather limited and still subject to controversy, and further research is needed to completely elucidate the role of biomarkers to predict plaque rupture and coronary events.23–25
Anatomic Distribution
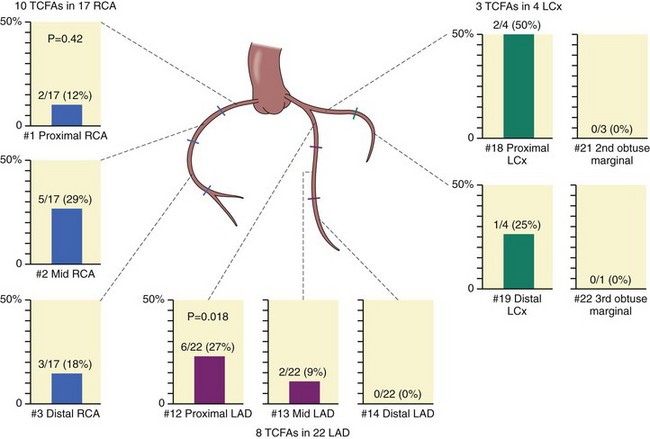
The anatomic distribution of coronary lesions responsible for AMI is dominated by the proximal segment, which is responsible for 80% of MIs in all three major vessels.26 These data were recently reproduced by OCT studies in vivo (Fig. 59-4).12,27,28 When located in bifurcations, TCFAs are predominately located in the proximal rim of the bifurcation, as evaluated by OCT and IVUS.29 After reviewing clinical characteristics, the next section provides the basis for understanding the pathophysiology of the disease. Most importantly, this section offers the foundation to critically evaluate novel imaging techniques that claim effectiveness in the diagnosis of high-risk VPs. For the interventionalist, the incidence of high-risk VPs evolving into clinical events is close to 13% in 3 years, in patients with ACS and multi-vessel disease. These lesions are positively remodeled, have large plaque burden, and are usually located in the proximal segment of the coronary arteries.
 Plaque Composition
Plaque Composition
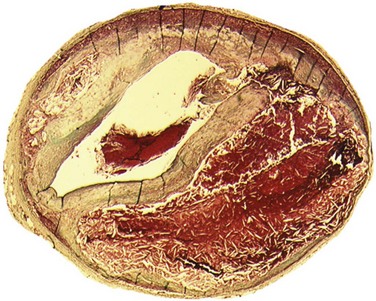
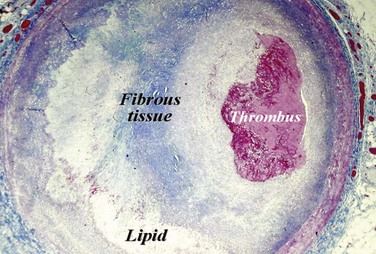
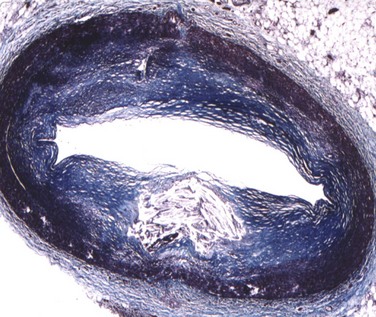
Plaque rupture is, by far, the most common cause of atherothrombosis, responsible for 70% to 75% of all events and up to 85% in hypercholesterolemic white males. In plaque rupture, disease progresses through lipid core expansion and macrophage accumulation at the edges of the plaque, leading to fibrous cap disruption (Fig. 59-5). As a result, identifying plaques at risk for rupture offers the possibility of preventing the most common substrate for coronary thrombosis. The second cause of atherothrombosis is plaque erosion. Included initially as “other causes of coronary thrombosis,” plaque erosion gained attention in the last decade as a significant substrate for coronary thrombosis and sudden cardiac death (SCD) in premenopausal females.13 As opposed to plaque rupture, erosion occurs in plaques with no specific features suitable for detection. Most of these plaques exhibit histologic patterns similar to plaques responsible for stable angina. They are characterized by a thick, smooth muscle cell–rich fibrous cap, reduced necrotic core areas, and a low degree of inflammation (Fig. 59-6). Plaque erosion is also associated with cigarette smoking, suggesting that thrombosis in these patients may be related to a systemic, prothrombogenic pathway rather than to a local, atherothrombotic mechanism. The characteristic lesion preceding plaque rupture is the TCFA, which is considered the hallmark of high-risk VPs (Fig. 59-7).30 The classic histologic patterns of TCFA include, but are not limited to, (1) a thin fibrous cap with increased stress–strain relationship; (2) large necrotic core with increased free cholesterol–esterified cholesterol ratio; (3) increased plaque inflammation; (4) positive vascular remodeling; (5) increased vasa-vasorum neovascularization; and (6) intra-plaque hemorrhage.31
Thin Fibrous Cap with Increased Stress–Strain Relationship
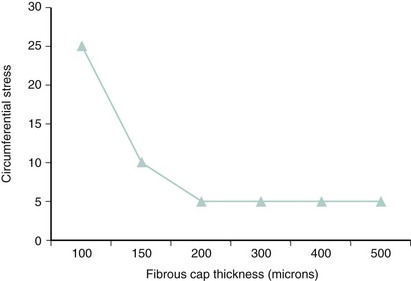
Autopsy studies have shown that ruptured plaques are characterized by a very thin fibrous cap, measuring 23 ± 19 microns (µm) in thickness. Most importantly, 95% of ruptured caps measured 64 µm or less in the coronary and 60 µm or less in the aorta.32 As a result, the first and probably most important histologic feature of TCFA is a fibrous cap 65 µm or more in thickness. These thin caps are unable to withstand the circumferential tensile stress applied by the oscillations of arterial blood pressure. The ratio of the circumferential tensile stress to the radial strain of the fibrous cap equals the stiffness of the tissue.33 Hence, soft (fatty) tissue will be more strained than stiff (fibrous) tissue when equally stressed. Furthermore, as caps become thinner, the stress increases in an exponential pattern (Fig. 59-8). In addition, as lipid pools become larger, stress also increases. Therefore, the strength of a cap may be as important as the actual thickness of a fibrous cap. This stress–strain relationship in the fibrous cap is therefore considered a feature for plaque vulnerability.34 Technology aiming to detect TCFA should have a radial resolution less than 65 µm and the ability to quantify the stress–strain relationship in the fibrous cap.
Large Necrotic Core with Increased Free Cholesterol to Esterified Cholesterol Ratio
Modified oxidized low-density lipoprotein (LDL) is avidly taken up by macrophages via scavenger receptors, leading to cytoplasm overload with lipid droplets. Continuous inflow of oxidized isoforms of LDL (oxLDL) leads to cell death with extracellular lipid accumulation within the matrix of the plaque. Preclinical experimentation has demonstrated how the oxLDL are associated with plaque vulnerability depending on the type of predominant molecule, mostly regulating apoptotic cell death.35,36 This is the basic mechanism of the necrotic core, which is formed after death by necrosis, or apoptosis, of lipid-laden macrophages, foam cells, and erythrocytes. Active collagen dissolution by metalloproteinases contributes to core expansion, which plays a major role in plaque vulnerability. In the aorta, TCFAs exhibit necrotic core areas of 40%, and ruptured plaques up to 50% of total plaque area.37 However, other studies in coronary arteries show lower necrotic areas, down to 24% and 34% in TCFAs and ruptured plaques, respectively.30 Core composition may influence the propensity for plaque rupture and thrombosis. An increased free cholesterol–esterified cholesterol ratio, with oxidized cholesterol, increases the likelihood of thrombosis by interacting with the oxLDL receptor-1 (LOX-1), and enhancing the expression of tissue factor.38–40
Increased Plaque Inflammation
Macrophages and T cells are capable of degrading the extracellular matrix by secretion of proteolytic enzymes such as plasminogen activators and matrix metalloproteinases (MMPs), including collagenases, elastases, gelatinases, and stromelysins, weakening the already thin fibrous cap and predisposing it to rupture.41 This has been recently confirmed by Suzuki et al, who found increased levels of MMP-1, MMP-13, and IL-6 in the coronary vessels of patients who had experienced MI, supporting the evidence behind the role of MMP in plaque rupture that leads to clinical events.42 Among the multiple MMPs identified to have a role in atherosclerosis, two particular subtypes, MMP-7 and MMP-12, seem more selectively localized in specific subsets of macrophages; especially, those located in the necrotic core have low arginase-I expression and represent a marker of activation and enhanced inflammation.43–45 The continuous entry, survival, and replication of monocytes or macrophages within plaques are aimed to remove oxLDL and reduce oxidation and reactive oxygen generation (ROS) products. In situations where the macrophage scavenger capacity is overloaded, cell death is activated by apoptosis, releasing MMPs and tissue factor.46 This link among inflammation, apoptosis, and thrombosis was elegantly documented by Hutter et al in human and murine atherosclerotic lesions.47 Therefore, plaque inflammation is a pivotal feature of plaque vulnerability.4,48 Recent studies have reported two different subclasses of monocytes or macrophages inside the atheroma.49 The first class of macrophages, classically known as activated macrophages, or M1, promote inflammation. The second subtype, identified as M2, appears to be anti-inflammatory. Therefore, the ratio between M1 and M2 macrophages has an impact on atherosclerosis progression, plaque regression, or both. It has been described that different factors shift this ratio toward an increased content of M2 cells, including increased T helper–secreted molecules, IL-4, IL-13, peroxisome proliferator-activated receptor delta (PPAR-δ) and the lipid sphingosin 1-phosphate (S1P).50–52 These M2 macrophages are also responsible for efferocytosis, or the removal of short-lived apoptotic bodies from the atheroma. Defective efferocytosis leads to secondary necrosis, a process linked to advanced disease. The failure of macrophage efflux, prothrombotic factors, inflammatory chemokines, and collagenases increase the necrotic core, leading to plaque rupture and thrombosis.53,54
Degree of Vascular Remodeling
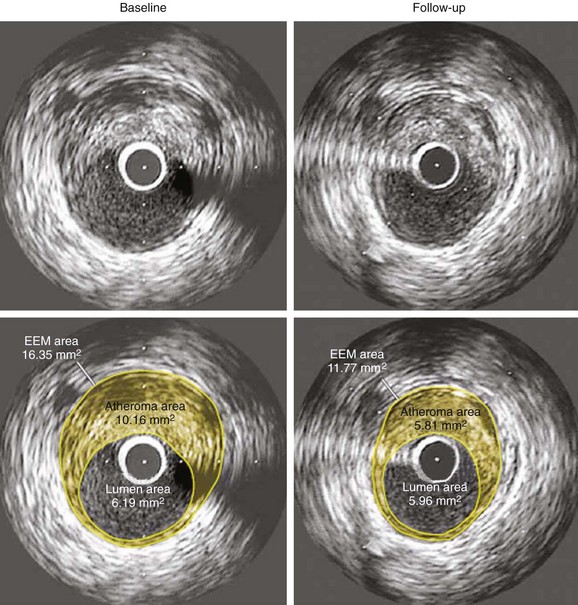
As a defense mechanism, the vessel wall can expand significantly to harbor large atheromas without obstructing the lumen (Fig. 59-9). Also known as remodeling, this process is linked to high-risk atherosclerosis. The mechanisms responsible for remodeling involve an inflammatory process at the base of the plaque, which leads to the digestion of the internal elastic lamina (IEL) and involves the deeper layers of the vessel wall, including the tunica media and the adventitia. Several studies have shown increased expression of MMPs within the intimo-medial interface of remodeled plaques.55,56 Furthermore, disruption of the IEL is associated with medial and adventitial inflammation, and evidence suggests that MMP-10 plays a regulatory role in atherosclerosis progression by influencing plaque inflammation and vascular remodeling.32,57 Concordantly, Burke et al demonstrated that marked expansion of the IEL occurred also in plaque hemorrhage, with or without rupture.58 The clinical relevance of remodeling was pioneered by Schoenhagen et al, who studied 85 patients with unstable coronary syndromes and 46 patients with stable coronary syndromes by using IVUS before PCI.59 Remodeling ratio (RR) was defined as the area of the external elastic membrane at the lesion divided by the same area at the proximal reference site. Positive remodeling was defined as an RR greater than 1.05 and negative remodeling as an RR less than 0.95 (Fig. 59-10). The RR was higher at target lesions in patients with ACS than in patients with stable angina. As a result, positive remodeling was more frequent in ACS (51.8% vs. 19.6%), whereas negative remodeling was more frequent in stable angina (56.5% vs. 31.8%) (P = 0.001), confirming the histopathologic associations between plaque remodeling and vulnerability.59 New technology aiming to detect TCFAs should provide exact measurements to quantify the degree of vascular remodeling. Of clinical relevance, Corti et al were the first to document the same eccentric pattern for plaque regression after aggressive lipid therapy.60 More recently, multiple studies have confirmed this observation (Figure 59-11).61–63 Considering that lipid is the main plaque component that can be reversed with therapy, this eccentric pattern of plaque regression suggests an effective reverse lipid transport system through the deeper layers of the vessel wall, probably mediated by vasa-vasorum neovascularization.4,64
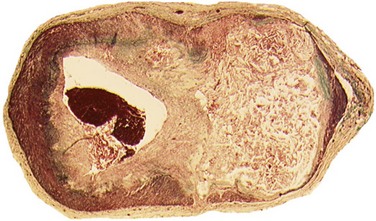
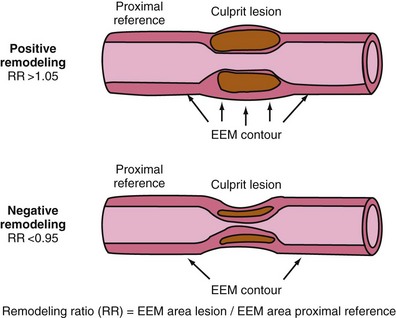
Figure 59-10 Cartoon explaining the direction of positive and negative remodeling. (See text for details.)
(Adapted with permission from Schoenhagen P, Ziada KM, Kapadia SR, Crowe TD, Nissen SE, Tuzcu EM: Extent and direction of arterial remodeling in stable versus unstable coronary syndromes: An intravascular ultrasound study, Circulation 101:598–603, 2000.)
Vasa-Vasorum Neovascularization
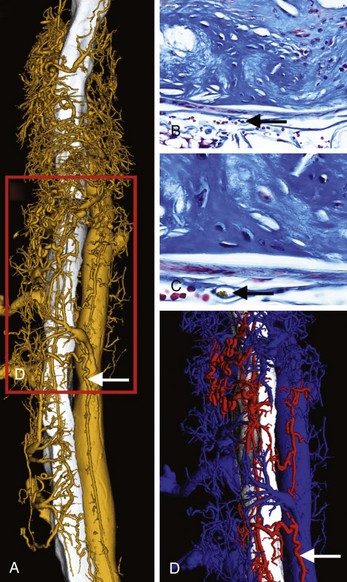
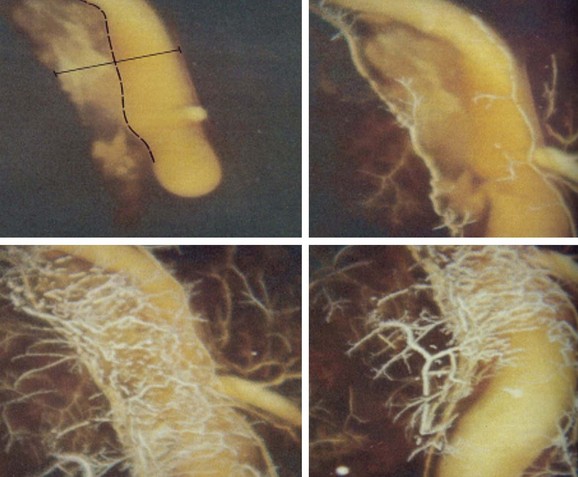
Atherosclerotic neovascularization evolves in early atherogenesis as a defense mechanism against hypoxia and oxLDL deposition within the tunica intima.65,66 In advanced disease, neovessels may play a defensive role allowing for lipid removal from the plaque through the adventitia leading to plaque regression, as described above. The adventitial vasa-vasorum is the main source of neovascularization in atherosclerotic lesions (Fig. 59-12). Neovascularization, elegantly delineated by Barger et al, who used cinematography (Fig. 59-13), is distributed from the epicardial fat to the plaque throughout vessel wall, although some can originate directly from the vessel lumen.67 Nevertheless, neovessels from the adventitial vasa were 28 times more numerous (96.5%) compared with those from the luminal side (3.5%).68 Neovessels from the adventitial vasa characterized severely stenotic lesions and correlated with the extent of inflammatory cell infiltration and lipid core size. Conversely, neovessels from a lumen origin were found in plaques with 40% to 50% stenosis and were associated more often with intra-plaque hemorrhage or hemosiderin deposits.68 Neovessels may also serve as a pathway for leukocyte recruitment to high-risk areas of the plaque, including the cap and the shoulder. Expression of vascular cell adhesion protein 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and E-selectin is twofold to threefold higher on neovessels compared with the arterial luminal endothelium, which confirms the pivotal role of neovessels as a pathway for leukocyte recruitment in human coronary plaques.69 Angiogenesis is stimulated by inflammatory cells. For instance, apoptotic microvesicles at the submicron level, found in atherosclerosis plaques, are highly proangiogenic by regulating CD40L and are produced mostly from macrophages.70,71 Histologic evidence for atherosclerotic neovascularization as a pathway for macrophage infiltration in advanced, lipid-rich plaques is also documented (Fig. 59-14).72 Neovessel content was significantly increased in plaques with severe inflammation, associated with both increased macrophage and T lymphocyte infiltration.73,74 Moreover, ruptured plaques exhibited the highest degree of neovascularization.75 Further analysis of plaque angiogenesis in diabetes documented a complex morphology, including sprouting, red blood cell (RBC) extravasation, and perivascular inflammation.76 Lastly, neovascularization may increase calcification, as demonstrated by the close link between immature endothelial cells and osteoblasts in atherosclerotic plaques and in cellular tissue, expressing vascular calcification–associated factor (VCAF).77 Using micro-CT imaging, recent studies have shown increased vasa-vasorum density, along with iron and glycophorin-A content in nonstenotic, noncalcified plaques. Moreover, calcium content was inversely proportional to neovessel content.78 Technology detecting TCFAs should quantify vasa-vasorum neovascularization in the adventitia, in the tunica media, and within the atherosclerotic plaque.
Intra-plaque Hemorrhage
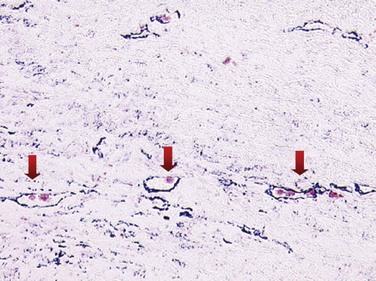
Neovessel leakage leads to extravasation of plasma content and RBCs into the plaque, which is known as intra-plaque hemorrhage (IPH). The mechanisms by which IPH occurs involves the leakage of RBCs from immature neovessels, which is mediated by various growth factors and chemokines that are expressed more in VPs.79 Compared with nonhemorrhagic atheromas, evidence has demonstrated lower levels of vascular endothelial growth factor (EGF), placental growth factor (PGF), and angiopoietin-1, in combination with an increased vascular endothelial growth factor (VEGF) expression, which are characteristic of hemorrhagic plaques.80,81 Lysis of RBCs contributes to lipid deposition. In addition, extracorpuscular hemoglobin (Hb) can induce oxidative tissue damage by virtue of its heme iron, with subsequent production of reactive oxygen species.79 The defense mechanism against free Hb is haptoglobin (Hp), which irreversibly binds to free Hb forming an Hp–Hb complex. Two classes of alleles (Hp-1 and Hp-2) characterize the human Hp locus at chromosome 16q22. The protein products of the two Hp alleles are structurally different, and the cardiovascular effects of this Hp polymorphism play a major role in patients with DM.82–84 Multiple independent epidemiologic studies examining incident cardiovascular disease have demonstrated that individuals with DM and the Hp 2-2 (homozygous for the Hp 2 allele) genotype have the risk of cardiovascular events four to five times higher compared with individuals with the Hp 1-1 (homozygous for the Hp 1 allele) genotype. The mechanism by which this Hp 2-2 phenotype regulates inflammation and enhances plaque vulnerability is related to decreased Hp clearance in the Hp 2-2 phenotype. This is explained, in part, by a defective CD163 receptor in macrophages, which is the receptor in charge of removing the Hp–Hb complex from the atheroma.85 Technology detecting TCFAs should identify intra-plaque hemorrhage, iron deposition, RBC membranes, and hemosiderin deposits in macrophages. In patients with DM, Ha genotyping may offer additional prognostic value.
 Plaque Imaging
Plaque Imaging
Invasive Techniques
Coronary angiography has the ability to delineate the coronary lumen, but it does not provide any information about the vessel wall. Therefore, this technique is not appropriate for the detection of VPs. Thus multiple intracoronary imaging techniques have been proposed to identify TCFAs. Of pivotal importance, every imaging technique should have appropriate validation, with histology as the gold standard to identify key histo-morphologic components. Considering that the majority of atherosclerotic plaques (TCFAs and non-TCFAs) will have a certain degree of fibrous cap thickness, shear stress patterns, necrotic core area, macrophage area, positive remodeling, and vasa-vasorum neovascularization, the presence or absence of these features alone (sensitivity and specificity) is not enough. Proper histologic validation must include accurate assessment of the degree of these components, which involves linear regression analysis. This validation process should be confirmed in animal models of TCFA before being applied in human coronary arteries.86 Then, the ultimate test should be a natural history study of all these techniques to determine if specific plaque components have any prognostic implications. Novel intracoronary techniques to detect TCFAs include (1) IVUS, (2) virtual histology (VH), (3) palpography, OCT, (4) IVUS elastography, or (5) intravascular magnetic resonance imaging (MRI), (6) angioscopy, (7) spectroscopy, and (8) thermography. A summary of these techniques, the component detected, and the resolution or accuracy is presented in Table 59-1. The first seven are already available for use in clinical practice or are under active evaluation in humans. With regard to thermography and the noninvasive modalities, an evidence-based approach for the understanding of their clinical usefulness will be presented. The interventionalist must develop a critical approach to evaluating these novel techniques, understanding their potential, and, most importantly, discerning their multiple limitations before considering them for clinical use.
TABLE 59-1 Summary of Current Invasive Detection Technologies
| Technology | Component Detected | Resolution/Accuracy |
|---|---|---|
| Intravascular ultrasound (IVUS) | Remodeling, calcium | 100–250 µm |
| IVUS–Virtual Histology | Necrotic core, calcium, collagen | 480 µm |
| Optical coherence tomography | Necrotic core, fibrous cap thickness, macrophages | 5–20 µm |
| IVUS–Elastography | Plaque strain | 100–250 µm |
| Intravascular magnetic resonance imaging (MRI) | Necrotic core | 250 µm |
| Angioscopy | Surface appearance of the plaque | N/A |
| Spectroscopy | Necrotic core | N/A |
| Thermography | Metabolic activity of the plaque | 0.05° C accurate |
N/A, not applicable.
Intravascular Ultrasound
Unlike angiography, IVUS allows proper visualization of the disease in the vessel wall and provides cross-sectional and longitudinal images of atherosclerotic plaques in vivo.87 IVUS is based on transmitting and receiving high-frequency sound waves from tissue through a low-profile catheter (approximately 1 mm); reaching a radial resolution between 100 to 250 µm. IVUS is safe, quick, and easy. Most importantly, IVUS allows identification of hemodynamically significant lesions that may be underestimated by angiography, particularly in nonocclusive plaques with positive remodeling. In addition, IVUS delineates the degree of calcification, plaque burden, and the degree of arterial remodeling. It uses the amplitude of the backscattered ultrasound signal to differentiate highly echogenic components such as calcium and dense fibrous tissue from echolucent tissue, including lipid and necrotic core. However, it cannot clearly differentiate between fibrous and fatty plaques.88 As a consequence, it is accepted that grayscale IVUS, as an isolated technology, is not capable of distinguishing plaque types. Thus, the application of other imaging protocols and algorithms to IVUS, including integrated backscatter IVUS analysis and Virtual Histology (IVUS-VH), which is discussed below, may contribute to better identification of the different plaque components. Researchers have studied backscatter analysis that extracts frequency components of a signal buried in the original IVUS signal. The imaging signal from a small volume of tissue creates an integrated backscatter (IB) pattern.89 Several studies have reported on the IVUS characteristics of culprit lesions and the presence of multiple ruptured plaques in patients with acute coronary events.87,90 A recent study evaluated the long-term outcomes of VPs arbitrarily defined as plaques with rupture, lipid core, dissection, or thrombus by conventional IVUS during ACS both in culprit and nonculprit locations. Multiplicity of VPs in the nontarget vessels (HR 2.2; 95% confidence interval [CI] 1.4–3.4, P = 0.001) was the only independent predictor of long-term critical events. Finally, DM and ACS were significantly associated with the multiplicity of VP.91
The ability of IVUS to identify TCFAs can be summarized as follows:
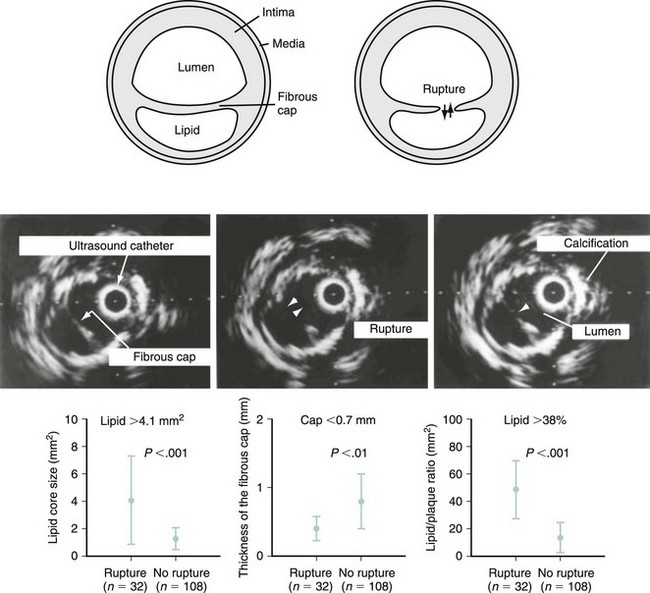
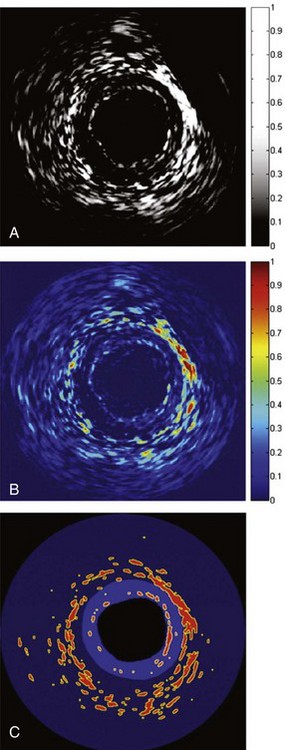
Figure 59-16 Differential intravascular ultrasound (IVUS) images to identify the vasa-vasorum, showing the subtracted post-injection signals from baseline signals. A, Black and white (signal intensity of Figure 59-1 A–C). B, Color-coded, panel A. C, Thresholded to show most significant areas of enhancement.
(Adapted with permission from Vavuranakis M, Kakadiaris IA, O’Malley SM, et al: Images in cardiovascular medicine. Detection of luminal-intimal border and coronary wall enhancement in intravascular ultrasound imaging after injection of microbubbles and simultaneous sonication with transthoracic echocardiography, Circulation 112:e1–e2, 2005.)
Virtual Histology
Considering the significant limitations of IVUS, Nair and Vince at Cleveland Clinic studied the ultrasound scattered reflection wave as a possible alternative to improve tissue characterization.107 This backscattered reflection wave is received by the transducer, where it is converted into voltage. This voltage is known as backscattered radiofrequency (RF) data. Using a combination of previously identified spectral parameters of the backscattered ultrasound signal, a classification scheme was developed to construct an algorithm to test plaque composition ex vivo. Four major plaque components were tested, including fibrotic tissue (dark green), fibro-fatty tissue (yellow-green), calcific-necrotic core (red), and dense calcium (white). A color was assigned for each of these components and is displayed on the IVUS image (Fig. 59-17). The Movat-stained histologic images identified homogeneous regions representing each of the four plaque components (Fig. 59-18). The unit of analysis (also called box) was initially composed of 64 backscattered RF data samples in length (480 µm).107 The algorithm developed was then validated ex vivo, with sensitivities and specificities between 79% and 93% for all four-plaque components.107 The initial studies were performed in ex vivo human coronary specimens with a 30-MGz, 2.9F, mechanically rotating IVUS catheter (Boston Scientific Corp); the initial catheter approved by the U.S. Food and Drug Administration (FDA) was a 20-MHz (Eagle-Eye® Gold) device. Recently, the catheter was upgraded with a 45-MHz transducer that is currently available for clinical practice. In vivo validation studies have shown positive results.108,109 Virtual histology gained significant attention with the PROSPECT Trial.15 As previously discussed in this review, VH-derived TCFA was associated with increased events (HR 3.35, P < 0.001). Most importantly, the highest-risk lesions (HR 11.05, P < 0.001) were a conglomerate of several features, including greater than 70% plaque burden, low MLA, and TCFA morphology. Therefore, isolating TCFA by using VH may not be enough to categorize a lesion as VP. The possibility of a PROSPECT II study randomizing patients with these lesions to aggressive medical therapy with or without stenting may be considered. However, the large number of patients needed and the increased costs may limit the ability to test this hypothesis.
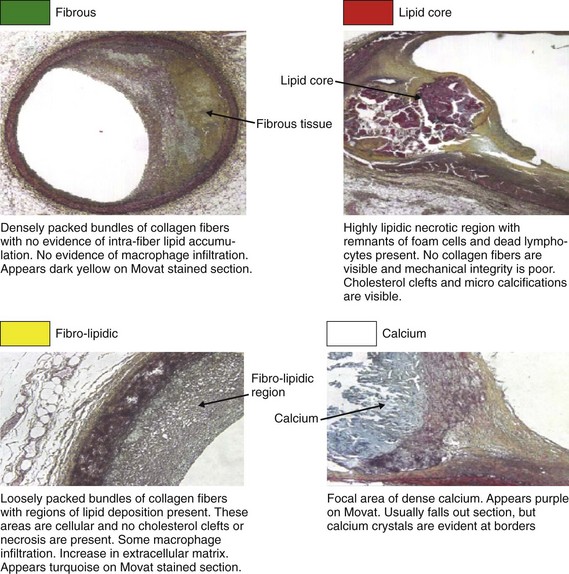
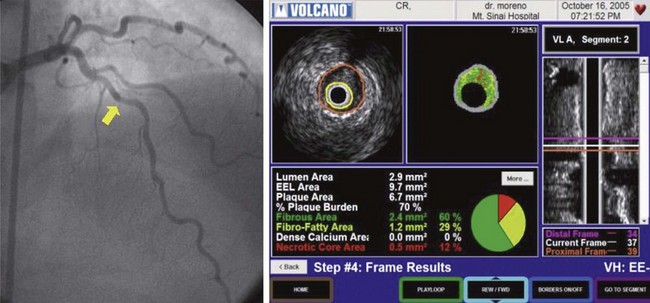
Figure 59-18 Definitions of the different plaque components obtained with intravascular ultrasound Virtual Histology (IVUS-VH).
(Reproduced with permission from Vince, DG, personal communication).
The individual ability of VH to identify TCFAs can be described as follows:
Elastography and Palpography
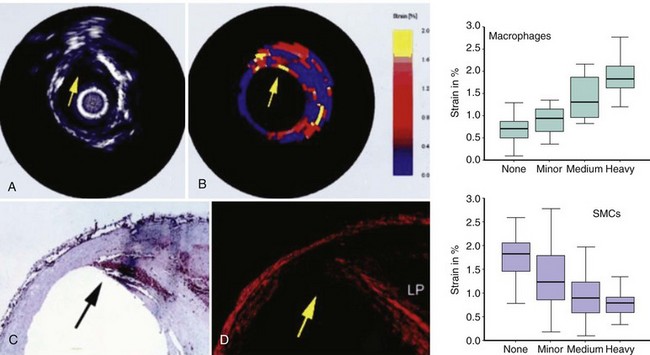
The mechanical stress induced by changes in blood pressure induces deformation on the fibrous cap (strain).127 This stress–strain relationship in coronary lesions can be identified by using another IVUS-derived technique called elastography or palpography, which displays data in a color-coded scale (see Figure 59-18).128 Purple indicates a low strain (hard, stiff), whereas yellow indicates a region of high strain (soft, deformable).129 Palpography has high sensitivity and specificity to detect VP, with a deformation of more than 2% reflecting increased macrophage infiltration, reduced smooth muscle cell, and low collagen content (Fig. 59-19).130 The Rotterdam Classification (ROC) divides strain into four subclasses; the worst is ROC IV, with a deformation greater than 1.2%.129 Palpography shows correlations with CRP, and ST elevation myocardial infarction (STEMI). Aggressive treatment using statins can reduce, over a period of 6 months, the intensity and frequency of these high-strain spots. Recent improvements using reconstructive compounding by motion artifact correction are promising.131,132 Nonetheless, palpography cannot detect fibrous cap thickness, necrotic core, degree of remodeling, neovascularization, or IPH.
Optical Coherence Tomography
OCT is a novel high-resolution intravascular imaging technique that is currently approved for clinical use. Of all of the invasive modalities, OCT provides the highest resolution (5 to 20 µm).133 Excellent sensitivity and specificity, between 92% and 100%, have been documented for all components of TCFA.133 Superb resolution allows for improved images (Fig. 59-20). Recent advances using optical frequency domain imaging (OFDI) allow for high-speed comprehensive imaging, scanning up to 5 cm with one single saline flush (Fig. 59-21).134 OCT is currently being used in clinical practice, providing significant information for the identification of plaque rupture, fibrous cap erosion, intracoronary thrombus, and TCFA location.135,136 Similarly, OCT can predict no-reflow phenomenon in patients with large lipid cores who undergo PCI during ACS.137 A direct comparison of OCT with IVUS-VH to detect TCFAs is shown in Figure 59-22. With all these promising facts on OCT, the interventionalist needs objective information regarding the ability of OCT to detect TCFA, which can be summarized as follows:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


