Fig. 6.1
Typical honeycomb lung. (a) High-resolution computed tomography (HRCT) shows clustered multiple cysts of which walls are relatively thick. The diameter of each cyst is one cm or less. Honeycomb lung is located subpleurally. (b) Reformatted coronal image shows a constellation of multiple cysts located in the subpleural regions
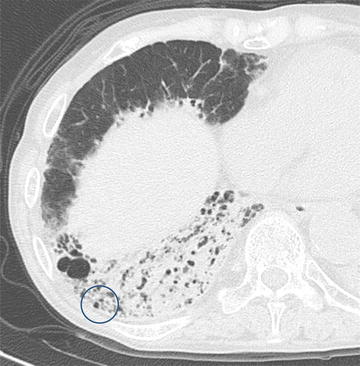
Fig. 6.2
Traction bronchiectasis in advanced fibrotic nonspecific interstitial pneumonia (fNSIP). High-resolution computed tomography (HRCT) of advanced fNSIP shows a constellation of traction bronchiectasis in the right middle lobe, evidenced by the course of the dilated bronchi within the CT plane. In the right lower lobe, a multicystic shadow may represent a constellation of traction bronchiectasis when dilated bronchi are sliced in the transverse plane perpendicular to the axes of the bronchi or honeycomb lung (circled)

Fig. 6.3
Pneumonia and emphysema (Swiss cheese appearance). (a) Chest x-ray shows overinflation of both lungs. Abnormal opacity is noted in the right middle to lower lung fields. (b) High-resolution computed tomography (HRCT) shows a multicystic shadow and ground-glass opacity/consolidation in the right lower lobe. Emphysema is evident in the lung fields without ground-glass opacity/consolidation
6.2.2 Disagreement Among Radiologists in Judging Honeycomb Lung
According to Fleischner Society nomenclature, radiological honeycomb lung describes findings of a cluster of relatively thick-walled cysts (3–10-mm diameter and 1–3-mm cyst wall thickness) in peripheral (subpleural) regions of the lung that are most frequently seen in IPF/UIP [4]. These radiological findings correspond with pathologic features – cystic space with small dilated airway and thick cyst wall with infolded fibrotic pulmonary parenchyma – that definitely differ from such destructive change as that of pulmonary emphysema.
Because the limited spatial resolution of HRCT prevents observation of microscopic honeycomb lung, radiological description must be confined to macroscopic honeycombing, and the term honeycomb lung must be used with extreme caution. Even experienced radiologists may disagree in judging honeycomb lung [5], primarily because various pathologic processes mimic its appearance and the term is used to describe different entities in the fields of pathology and radiology.
Mimickers of honeycomb lung in emphysema include pneumonia and other disease processes with diffuse ground-glass opacity (GGO)/consolidation, though GGO/consolidation does not occur in the holes of emphysema, pneumonia that resembles Swiss cheese on HRCT (Fig. 6.3), and the multiple cysts frequently observed in pulmonary fibrosis concomitant with emphysema (Fig. 6.4a, b).
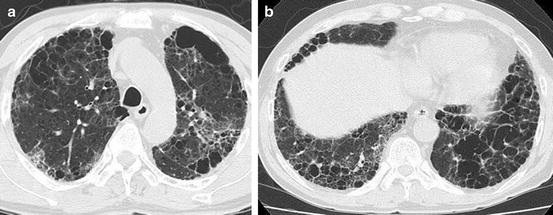
Fig. 6.4
Pulmonary fibrosis concomitant with pulmonary emphysema. (a) Computed tomography (CT) of upper lung shows paraseptal and centrilobular emphysema mixed with ground-glass opacity and reticular opacity. (b) CT scan of lower lung shows multiple cysts including large thick-walled cysts that mimic honeycombing
Constellated traction bronchiectasis may also simulate honeycomb lung when dilated bronchi are sliced in the transverse plane perpendicular to their axes (Fig. 6.2). The use of coronal/sagittal reconstructed images or 3-dimensional display may improve its differentiation from honeycombing. It is very confusing that traction bronchiectasis may intermingle with honeycombing and that a constellation of “pure” traction bronchiectasis may simulate honeycomb lung (Fig. 6.5a, b).
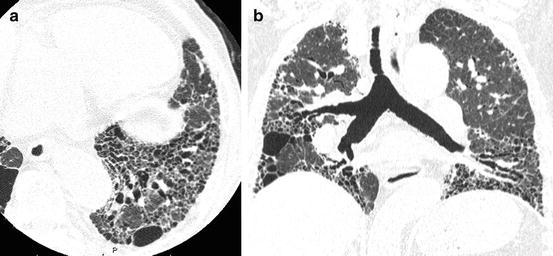
Fig. 6.5
Advanced fibrotic nonspecific interstitial pneumonia (fNSIP). (a) High-resolution computed tomography (HRCT) of lower lung shows traction bronchiectasis mixed with multiple cysts that mimics honeycomb lung. (b) Reformatted coronal image shows most of cystic structures are constellation of traction bronchiectasis
6.3 Imaging Findings of Idiopathic Pulmonary Fibrosis/Usual Interstitial Pneumonia
6.3.1 Pathologic Criteria of IPF/UIP
Pathologic hallmarks of the UIP pattern include perilobular/periacinar fibrosis and spatial and temporal heterogeneity. Advanced fibrotic change, such as honeycombing, exists in close proximity with nearly normal lung tissue, typically within the same pulmonary lobule, and the borders of fibrotic foci are very sharp and abruptly abut lung that is nearly normal [6].
Fibrosis with a UIP pattern may be seen in such diseases as secondary interstitial pneumonia of known cause, interstitial pneumonia with collagen vascular disease (CVD-IP), chronic hypersensitivity pneumonitis (CHP), sarcoidosis, and asbestosis. Pathologic findings of CVD-IP frequently show rich aggregated lymphatic follicles with germinal center, pleuritis, bronchiolitis, and other features. Typically, CHP shows airway-centered fibrosis and small granulomas related to airways, but these features may be absent. The multidisciplinary discussion to diagnose IPF/UIP must exclude these secondary IPs with a UIP pattern.
The 2011 international diagnostic guideline [1] classifies pathologic patterns as definite, probable, possible, and not UIP based on the combination of several pathologic findings.
A definite UIP pattern comprises marked fibrosis/architectural distortion with or without honeycomb lung located predominantly in subpleural and paraseptal regions, patchy involvement by fibrosis, fibroblastic foci, and absence of any finding that contraindicates the diagnosis of IPF/UIP. Findings that rule out IPF/UIP include hyaline membrane, organizing pneumonia, marked inflammatory cell infiltration, granulomas, predominant airway-centered change, and any other finding that suggests an alternative diagnosis.
A probable UIP pattern includes marked fibrosis/architectural distortion with or without honeycomb lung, absence of either patchy involvement or fibroblastic foci, and absence of any finding that contraindicates the diagnosis of IPF/UIP.
A possible UIP pattern demonstrates patchy involvement of the lung by fibrosis with or without honeycomb lung and absence of any finding that contraindicates the diagnosis of IPF/UIP.
The definite UIP pattern includes perilobular (subpleural and/or paraseptal)/periacinar fibrosis and temporal/spatial heterogeneity, but the probable and possible UIP patterns may include other pathologic patterns than those of IPF/UIP, such as fibrotic nonspecific interstitial pneumonia (fNSIP), interstitial pneumonia with collagen vascular disease, or chronic hypersensitivity pneumonia.
6.3.2 HRCT Criteria of IPF/UIP
HRCT images must be included in the analysis of radiological findings of interstitial pneumonia [7–10]. Nishimura and associates described HRCT findings of IPF/UIP as perilobular fibrosis (abnormal opacities along the pleura, bronchial wall, large vessels, and interlobular septa) [11] (Fig. 6.6). Intralobular interstitial changes correspond with periacinar fibrosis along the intralobular veins. Fibrotic change is shown by reticular opacity or honeycomb lung. Honeycomb lung is a key finding of IPF/UIP and more extensive in UIP than NSIP [12–17] (Figs. 6.5 and 6.6).
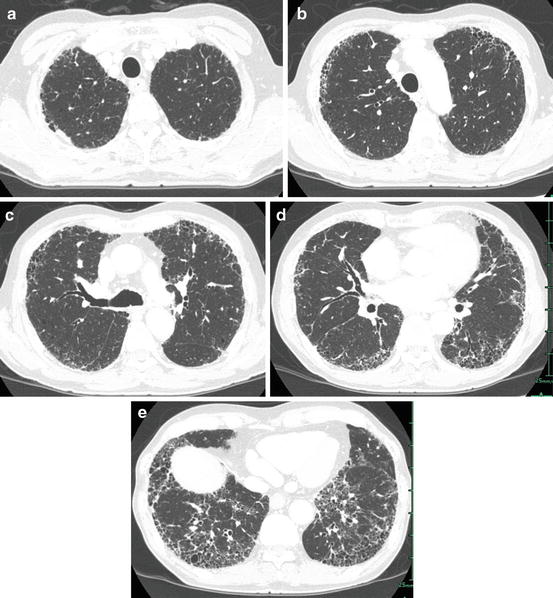
Fig. 6.6
Idiopathic pulmonary fibrosis/usual interstitial pneumonia (IPF/UIP). (a) High-resolution computed tomography (HRCT) at the pulmonary apex shows small linear opacity abutting the chest wall that suggests perilobular (pleural surface, interlobular septa, and bronchovascular bundle) fibrosis. (b) HRCT at the level of the aortic arch shows inhomogeneous reticular opacity abutting the chest wall (subpleural regions). (c) HRCT at the level of the tracheal bifurcation shows inhomogeneous reticular opacity and honeycomb lung abutting the chest wall. Small linear opacity adjacent to the chest wall is also identified. (d) HRCT at the level of the pulmonary vein shows subpleurally located reticular opacity and honeycomb lung. Distribution of abnormal opacity is patchy and confined to subpleural regions. (e) HRCT at the level of the lung base shows patchy areas of reticular opacity and honeycomb lung in the subpleural regions. No findings contraindicate the diagnosis of IPF/UIP
Approximately one third of UIP diagnosed by video-assisted thoracoscopic surgery (VATS) mimics NSIP at HRCT [18, 19], probably because surgical lung biopsy is performed to diagnose IPF/UIP with atypical findings on radiology and not disease with typical features (Fig. 6.7). IPF/UIP may show an NSIP pattern during the course of disease [20, 21], and a UIP pattern may change to an NSIP pattern in the terminal stage of disease. The pattern of pathologically proven NSIP comorbid with pulmonary emphysema mimics a UIP pattern at HRCT [22].
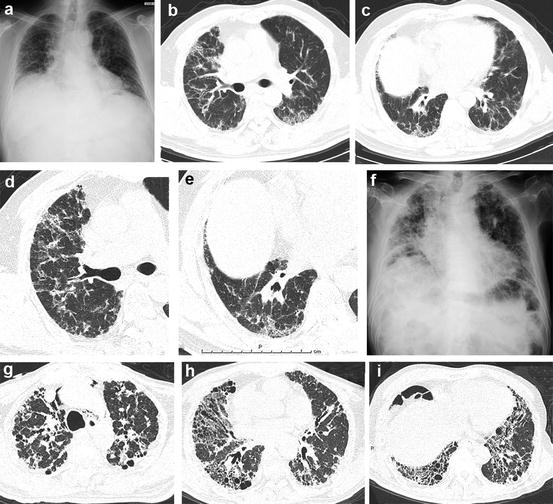
Fig. 6.7
Chronic fibrosing interstitial pneumonia of unknown cause with definite usual interstitial pneumonia (UIP) pattern at pathology and atypical (inconsistent with) UIP pattern at high-resolution computed tomography (HRCT). (a–g) Images at surgical lung biopsy; (f–i) images were obtained 4 years later. (a) Chest x-ray shows volume loss in bilateral lungs with reticular opacity. (b) HRCT at the middle lung shows ground-glass opacity (GGO) and reticular opacity in the peribronchovascular regions in bilateral lungs. (c) GGO and reticular opacity predominant in basal regions. Abnormal opacity is identified in both subpleural and peribronchovascular regions. (d) HRCT at the middle lung shows peribronchovascular abnormal opacity that represents a pattern inconsistent with UIP. (e) HRCT at the level of the lung base shows abnormal opacity in both the peribronchovascular and subpleural regions. (f) Chest x-ray after 4 years shows progression of abnormal opacity and loss of volume in bilateral lungs. (g) HRCT at the level of the pulmonary apex shows peribronchovascular and subpleural foci consolidation with traction bronchiectasis. (h) HRCT at the level of the middle lung shows peribronchovascular-predominant reticular opacity with traction bronchiectasis that suggests a pattern of fibrotic nonspecific interstitial pneumonia (fNSIP). (i) In the lung base, HRCT shows peribronchovascular-predominant reticular opacity that includes traction bronchiectasis and cysts
6.3.2.1 Categories of HRCT Patterns in the 2011 Guideline (Table 6.1)
The 2011 IPF/UIP guideline [1] categorizes the disease patterns of IPF/UIP based on HRCT criteria as definite UIP (Fig. 6.6), possible UIP (Fig. 6.8), and inconsistent with UIP (Fig. 6.7). Certainty of IPF/UIP diagnosis is achieved by combining the categories of HRCT and pathology.
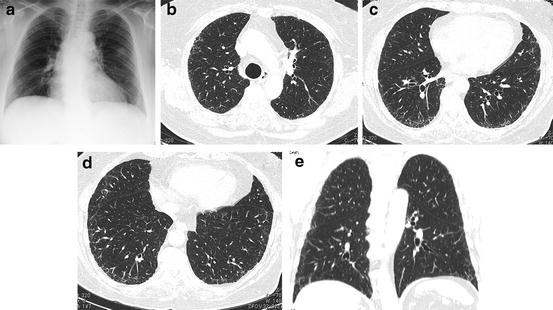
Table 6.1
High-resolution computed tomography (HRCT) categories of disease patterns of usual interstitial pneumonia (UIP) of the 2011 international guideline for the diagnosis and treatment of idiopathic pulmonary fibrosis [1]
HRCT criteria for UIP pattern | ||
|---|---|---|
UIP pattern (all four features) | Possible UIP pattern (all three features) | Inconsistent with UIP pattern (any of the seven features) |
Subpleural, basal predominance | Subpleural, basal predominance | Upper or mid-lung predominance |
Reticular abnormality | Reticular abnormality | Peribronchovascular predominance |
Honeycombing with or without traction bronchiectasis | Absence of features listed as inconsistent with UIP pattern (see third column) | Extensive ground-glass abnormality (extent > reticular abnormality) |
Absence of features listed as inconsistent with UIP pattern (see third column) | Profuse micronodules (bilateral, predominantly upper lobes) | |
Discrete cysts (multiple, bilateral, away from areas of honeycombing) | ||
Diffuse mosaic attenuation/air trapping (bilateral, in 3 or more lobes) | ||
Consolidation in bronchopulmonary segment(s)/lobe(s) | ||
Fig. 6.8
Possible usual interstitial pneumonia (UIP) pattern at high-resolution computed tomography (HRCT). (a) Chest x-ray shows ground-glass opacity predominantly in bilateral basal lungs. Nodular opacity in the left middle lung field suggests lung cancer. (b) HRCT at the upper lung shows subpleural inhomogeneous reticular opacity. (c) HRCT at the lower lung shows subpleurally located inhomogeneous reticular opacity. Reticular opacities distribute inhomogeneously. (d) HRCT at the lung base shows subpleurally distributed reticular opacity without honeycomb lung. (e) Coronal CT shows subpleural reticular opacity located predominantly in the lower lungs. No honeycombing is identified
A definite IPF/UIP pattern at HRCT consists of subpleural distribution of reticular opacity or honeycomb lung predominantly in the dorsal aspects of the lower lobes and absence of any of the below mentioned 7 findings noted that contraindicate a diagnosis of IPF/UIP. A possible IPF/UIP pattern demonstrates lower lobe predominance and subpleural distribution, reticular opacity, and lack of any of the 7 findings that contraindicate a diagnosis of IPF/UIP; the honeycombing of the definite IPF/UIP pattern is absent. A pattern that is inconsistent with IPF/UIP includes any of the 7 findings that contraindicate IPF/UIP, peribronchovascular-predominant distribution (Fig. 6.9), prominent GGO (wider than reticular opacity and honeycomb lung) (Fig. 6.10), upper lung predominance as pleuropulmonary fibroelastosis (PPFE) (Fig. 6.11), a large cyst away from honeycomb lung (Fig. 6.12), segmental distribution of shadow, profuse micronodules (Fig. 6.13), and diffuse mosaic appearance. These imaging findings may represent secondary interstitial pneumonias with a UIP pattern that may be included in the differential diagnosis of IPF/UIP.
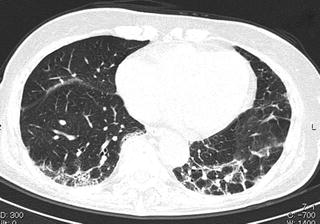
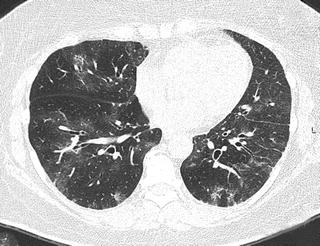
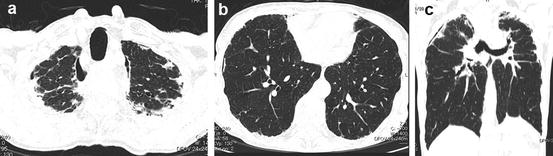
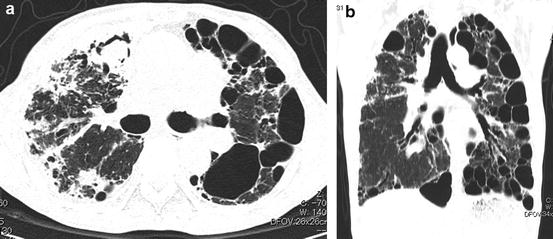
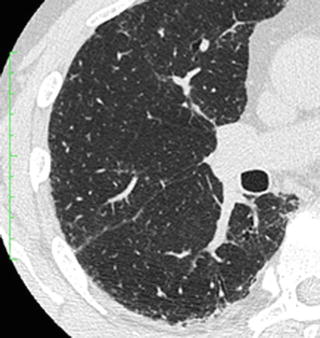
Fig. 6.9
Interstitial pneumonia in a patient with dermatomyositis. Prominent peribronchovascular distribution. High-resolution computed tomography (HRCT) shows peribronchovascular consolidation that suggests acute lung injury (fibrosing organizing pneumonia [OP])
Fig. 6.10
Cellular nonspecific interstitial pneumonia (NSIP) and prominent ground-glass opacity (GGO). High-resolution computed tomography (HRCT) shows widespread GGO without structural distortion that suggests cellular NSIP
Fig. 6.11
Pleuroparenchymal fibroelastosis (PPFE). (a) High-resolution computed tomography (HRCT) at the pulmonary apex shows subpleural atelectatic fibrosis. (b) HRCT at the pulmonary base shows subpleural fibrosis that suggests a usual interstitial pneumonia (UIP) pattern. (c) Reformatted coronal CT image shows marked volume loss in bilateral upper lobes with elevation of the pulmonary hili
Fig. 6.12
Chronic hypersensitivity pneumonia with large cyst. (a) High-resolution computed tomography (HRCT) image shows multiple large subpleural cysts. Fungal ball is noted in the right lung. (b) Reformatted coronal images show multiple large cysts in the subpleural region
Fig. 6.13
Profuse micronodules of respiratory bronchiolitis interstitial lung disease (RBILD). High-resolution computed tomography (HRCT) shows prominent centrilobular nodules
In IPF/UIP, HRCT findings of overlapping reticular and ground-glass opacity (GGO) combined with honeycombing demonstrate a wider area of fibrosis than GGO. Prominent GGO suggests less fibrotic change. Profuse micronodules are frequently seen in chronic hypersensitivity pneumonia and sarcoidosis, and peribronchovascular-predominant abnormal opacity is typically seen in fibrotic NSIP. Absence of segmental consolidation is intended to exclude pneumonia. A large cyst is typically seen in complicated emphysema or cystic diseases, and predominant upper distribution is a hallmark of such inhalational diseases as pneumoconiosis and CHP. Diffuse mosaic appearance suggests airway disease, sometimes identified in CHP.
6.3.2.2 Correlation of HRCT and Pathologic Findings (Figs. 6.6 and 6.8)
The 2011 diagnostic guideline for IPF/UIP [1] describes limited specific findings on HRCT and none that reflect temporal/spatial heterogeneity, and other forms of interstitial pneumonia, such as fibrotic NSIP (fNSIP), may manifest a pattern of possible UIP on HRCT. Pathologic hallmarks of the UIP pattern are perilobular/periacinar fibrosis and temporal/spatial heterogeneity. Fibrosis is assured by the presence of reticular opacity and/or honeycombing at HRCT, and perilobular fibrosis corresponds with the subpleural location of reticular opacity or honeycomb lung.
Though few articles in the English literature describe the heterogeneous CT appearance of IPF/UIP, HRCT findings that reflect the heterogeneous distribution of fibrosis must be investigated [23]. Potentially useful findings that suggest heterogeneity may include marked laterality of abnormal opacity, coexistence of advanced fibrosis (such as honeycombing) with normal-appearing lung parenchyma packed in a very small area, and a normal-appearing area remaining within subpleural reticular opacity and/or honeycomb lung [24, 25].
Approximately one third of VATS-determined IPF/UIP shows an atypical CT appearance that mimics NSIP or unclassified patterns (which might represent secondary UIP), probably because surgical biopsy was not performed in recent cases of IPF/UIP that demonstrated a typical IPF pattern (definite UIP pattern) [18]. However, the prognosis is similar for IPF/UIP with atypical findings and IPF with a definite IPF/UIP appearance at HRCT [19].
6.3.2.3 Honeycomb Lung in the Diagnosis of IPF/UIP (Table 6.1, Fig. 6.14)
Honeycomb lung is one of the most important findings of IPF/UIP [16, 26–29] and can be key in its diagnosis [1]. Definitive diagnosis of IPF/UIP is possible without surgical biopsy when HRCT shows a typical UIP pattern, but biopsy is necessary when imaging demonstrates a possible or inconsistent with UIP pattern. Honeycombing is the only finding different between the patterns of definite and possible UIP, and difficulty in its discrimination on radiology findings, even among experienced chest radiologists [5], requires that surgical lung biopsy be performed. Heavy reliance on the observation of honeycombing to establish the diagnosis of IPF/UIP therefore can be problematic.
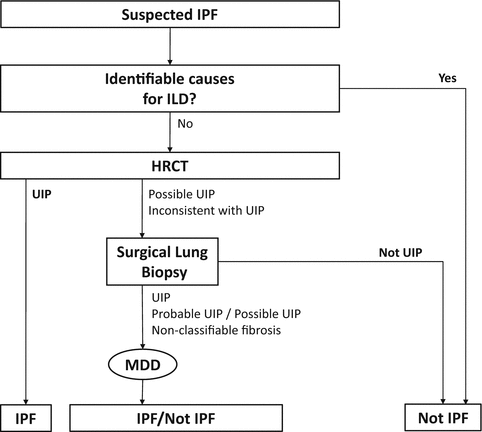
Fig. 6.14
Approach of the 2011 international guideline for the diagnosis of idiopathic pulmonary fibrosis [1]
6.3.2.4 Diagnosis of IPF/UIP in Early Stage
New drugs have recently been developed to treat IPF/UIP, and medical intervention might be more effective in the early stage of disease prior to honeycombing. Pathologic findings of IPF/UIP indicate that perilobular/periacinar fibrosis and temporal/spatial heterogeneity should be included in the diagnostic criteria of HRCT. Early recognition of these features in the disease process could improve treatment management.
6.4 Differential Diagnosis of IPF/UIP
6.4.1 Nonspecific Interstitial Pneumonia
Satisfactory differentiation of subtypes of idiopathic interstitial pneumonias by HRCT has been reported generally [30]. NSIP is one the most important differential diagnoses of IPF/UIP [31, 32]. Its primary HRCT feature is peribronchovascular or patchy ground-glass opacity. Findings of intralobular reticular opacity, traction bronchiectasis within GGO, and honeycomb lung indicate progression of fibrosis and suggest a worse prognosis (Fig. 6.15) [17, 33–41]. According to Sumikawa and associates, IPF/UIP shows more extensive honeycombing than fNSIP [12].
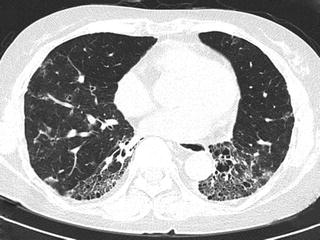
Fig. 6.15
Fibrotic nonspecific interstitial pneumonia (fNSIP). High-resolution computed tomography (HRCT) at the lower lung shows marked volume loss in bilateral lower lobes. Reticular opacity and traction bronchiectasis are identified in bilateral lower lobes
Approximately one third of VATS-determined IPF/UIP appears atypical on HRCT, but the prognosis is similar for IPF/UIP with typical imaging features and disease with atypical features that requires surgical lung biopsy [19].
6.4.2 Desquamative Interstitial Pneumonia
HRCT findings of desquamative interstitial pneumonia (DIP) include subpleural or patchy panlobular ground-glass opacity. Usually reticular opacity is less prominent, and honeycomb lung is absent. Multiple cystic changes are observed within areas of GGO, especially after disappearance of GGO by treatment [42]. During long-term follow-up study, honeycombing can become apparent [43].
6.4.3 Respiratory Bronchiolitis Interstitial Lung Disease (Fig. 6.13)
Respiratory bronchiolitis interstitial lung disease (RBILD) is another form of idiopathic interstitial pneumonia closely related to cigarette smoking [44]. Its main feature on HRCT is abundant centrilobular nodules that reflect respiratory bronchiolitis; other findings include patchy ground-glass opacity and reticular opacity [45, 46].
6.4.4 Sarcoidosis
Pulmonary sarcoidosis may mimic IPF at HRCT [47]. Profuse micronodules with perilymphatic distribution suggest the diagnosis of sarcoidosis.
6.4.5 Chronic Hypersensitivity Pneumonia (Fig. 6.16)
Chronic hypersensitivity pneumonia is a secondary interstitial pneumonia that shows a UIP or NSIP pattern on pathologic specimens [48]. Its imaging features most frequently show a UIP pattern but include upper lobe predominance or generalized distribution in the craniocaudal direction, profuse centrilobular micronodules in the upper lungs that suggest bronchocentric fibrosis, bridging fibrosis that connects the centrilobular and perilobular regions, cyst formation in the upper lungs, atelectatic induration, and/or prominent ground-glass opacity [49]. Some CHP cases simulate IPF/UIP, lacking any findings contraindicating this diagnosis, so the diagnosis of CHP requires multidisciplinary discussion among specialists [50, 51].
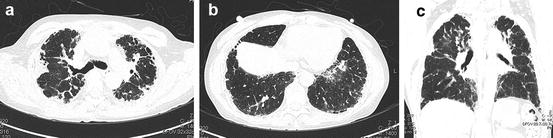
Fig. 6.16
Chronic hypersensitivity pneumonitis (CHP). (a) High-resolution computed tomography (HRCT) at the pulmonary apex shows multiple subpleural areas of atelectatic induration abutting the chest wall. Small cysts are also identified. (b) HRCT of the lower lung shows interstitial pneumonia with a UIP pattern. (c) Coronal reformatted image shows that reticular opacity is confined to subpleural regions
6.4.6 Collagen Vascular Disease (CVD) (Fig. 6.17)
Interstitial lung disease in patients with collagen vascular disease (CVD) most frequently shows an NSIP pattern, but a UIP pattern may be seen, especially in patients with rheumatoid arthritis [52–54]. Features of usual interstitial pneumonia with CVD (CVD-UIP) are somewhat atypical from those of IPF/UIP, and concomitant peribronchovascular abnormal opacity in addition to subpleural reticular opacity and/or honeycomb lung indicate concomitant UIP and NSIP patterns. On the other hand, Assayag’s group described no difference between HRCT images of UIP complicated with rheumatoid arthritis and those of IPF/UIP [55].
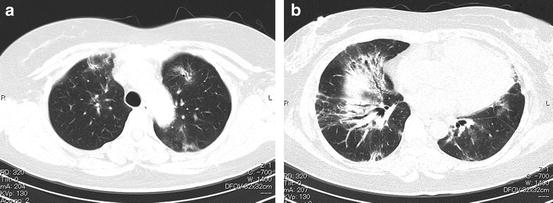
Fig. 6.17
Interstitial pneumonia with collagen vascular disease (CVD-IP) (fibrosing nonspecific interstitial pneumonia [NSIP] pattern). (a, b) High-resolution computed tomography (HRCT) images show abnormal opacities located predominantly in the lower lungs that represent peribronchovascular ground-glass opacity and consolidation including traction bronchiectasis
Because NSIP is frequently complicated in patients with CVD, Kinder and colleagues suggested NSIP might be a lung complication in patients with undifferentiated connective tissue disease (UCTD) [56]. Though the prognoses differ distinctly between patients with UCTD and those with usual NSIP [57], no differences in HRCT findings have been reported.
Lung-dominant connective tissue disease (LD-CTD) [58] and autoimmune-featured interstitial lung disease (AFILD) [59] are reported to be interstitial pneumonias that do not satisfy the diagnostic criteria of specific collagen disease but have clinical and/or pathologic findings that suggest CVD.
Though CVDs most frequently complicate NSIP, approximately half of AFILD shows a UIP pattern. Some LD-CTD/AFILD shows an unclassifiable (mixed UIP and NSIP pattern) or UIP pattern at HRCT. Because interstitial pneumonia in patients with UCTD, LD-CTD, and AFILD is currently classified as idiopathic interstitial pneumonia, IPF/UIP may include these interstitial pneumonias with the “flavor of CVD.”
6.4.7 Pneumoconiosis
Asbestosis is bronchocentric fibrosis caused by asbestos fiber (2 or more asbestos bodies/one square cm in histologic section). Bronchocentric fibrosis extends to the perilobular regions and may show the fibrosis of a UIP pattern [60–62].
Imaging findings of asbestosis differ by disease stage. Early stage findings include small nodular centrilobular opacities that correspond with bronchocentric fibrosis, thickening of the intralobular interstitium, septal thickening, subpleural curvilinear opacity that corresponds with coalescent bronchocentric fibrosis, wedge-shaped abnormal opacity abutting the chest wall, and air trapping [63]. Advanced asbestosis shows honeycomb lung and atelectatic fibrosis [64–66].
Although asbestosis may show fibrosis with a UIP pattern [67], typical HRCT images of classical asbestosis can show centrilobular nodules, subpleural curvilinear opacity, transpulmonary band, and prominent air trapping and less frequent honeycomb lung and traction bronchiectasis than IPF/UIP [68].
Pleural plaque on chest x-ray (CXR)/CT and clinical history of asbestos exposure are also useful in diagnosing asbestosis. Mixed dust pneumoconiosis and hard metal lung may show a UIP pattern [69].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


