Fig. 20.1
Skin. KS showing epidermis on the left and proliferation of spindle cells with many vascular channels and mild inflammatory reaction on the mid and right side of the field
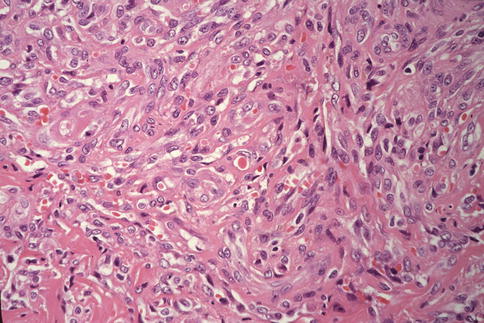
Fig. 20.2
KS, closer view. The lesion shown here consists of intersecting fascicles of spindle cells and proliferation of small blood vessels. Note single red blood cells within some capillary lumina
Among patients with KS, about one third have clinically evident pulmonary disease, and one half have pulmonary involvement at autopsy (Kornfeld and Axelrod 1983; Ognibene et al. 1985; Meduri et al. 1986; Zibrak et al. 1986; Garay et al. 1987; Fouret et al. 1987). Endobronchial lesions can be seen in about 50 % of patients (Pitchenik et al. 1985; Greenberg et al. 1985; Meduri et al. 1986; Zibrak et al. 1986; Garay et al. 1987; Fouret et al. 1987; O’Brien and Cohn 1989; Nathan et al. 1990). Clinical pulmonary manifestations are shortness of breath, fever, dry cough, hemoptysis, and chest pain. Some patients have no symptoms but may present with an abnormal chest radiograph. Among patients with KS who present with a respiratory problem, up to 50 % have parenchymal involvement (White and Matthay 1989). KS can also involve the airways, the pleura, and intrathoracic lymph nodes (Farber and Mark 1990; Brusch and Mark 1993). At autopsy, the lungs show multiple small scattered hemorrhagic nodules that are visible on the pleural surface and in the bronchial mucosa (Fig. 20.3). On section, the nodules may appear in scattered or aggregated forms (Fig. 20.4). Histologically, the lung lesions are similar to those seen in other organs. Low-power views show an infiltrative growth along the lymphatic vessels, mainly into the interstitium, the interlobular septa, around the pulmonary arteries, and bronchus and bronchioles (Fig. 20.5). The lesions consist of spindle cells mixed with inflammatory cells and irregular vascular channels (Fig. 20.6).
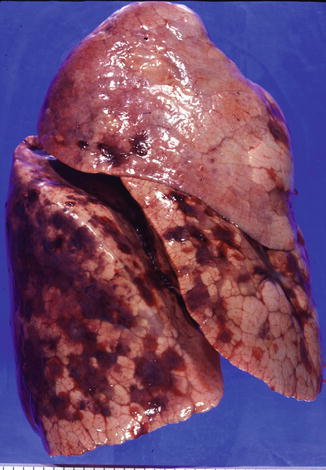
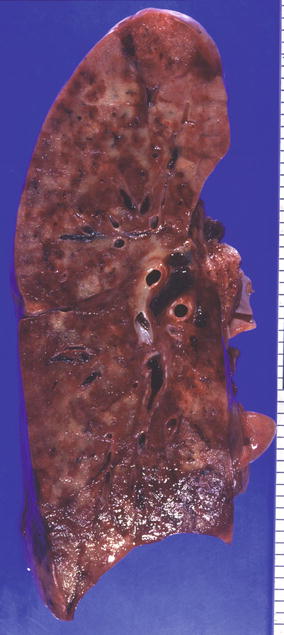
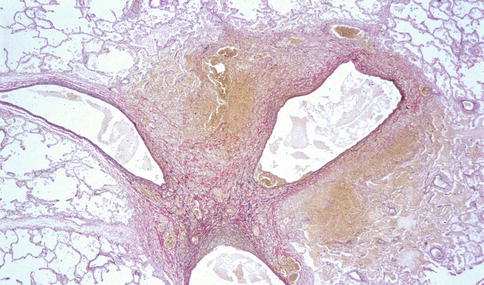
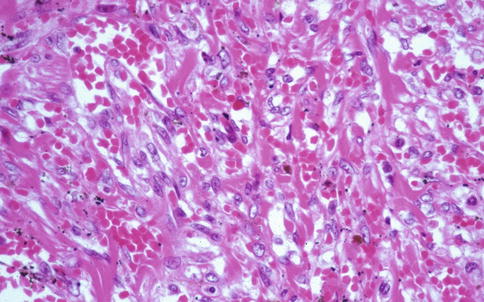
Fig. 20.3
KS in lung, gross appearance. An autopsy lung shows multiple small hemorrhagic nodules on the pleural surface (Courtesy of Dr. Morio Koike)
Fig. 20.4
KS in lung cross sections. Scattered yellow-white nodules and diffuse hemorrhage (Courtesy of Dr. Morio Koike)
Fig. 20.5
The pulmonary KS lesions extend into the interlobular septa, around the pulmonary arteries and bronchioles. Low-power view. Elastic stain (Courtesy of Dr. Morio Koike)
Fig. 20.6
Microscopically, pulmonary KS lesions consist of spindle cells, irregular vascular channels, and scant inflammatory cells, not too different from those seen in cutaneous lesions (Courtesy of Dr. Morio Koike)
Treatment is primarily palliative. Localized variants of KS can be successfully treated surgically. Highly active antiretroviral therapy (HAART) has proven very useful in the management of the disease (Berson et al. 1990; Conant et al. 1997; Murphy et al. 1997; De Milito et al. 1999). Radiation therapy has been successful in selected cases. Treatment against HIV infection usually improves and sometimes entirely eliminates KS in AIDS patients. Prevention depends on avoiding infection and maintaining a healthy immune system. A vaccine is currently not available.
20.3.2 Primary Effusion Lymphoma
Primary effusion lymphoma (PEL) is a distinct lymphoid neoplasm of B-cell lineage. Clinically, PEL presents as a malignant effusion in the pleura, pericardium, and peritoneum, without a detectable or discrete tumor mass. PEL is universally associated with HHV-8. PEL occurs in elderly men and mostly in areas with a high prevalence of HHV-8, such as the Mediterranean basin. Primary effusion lymphoma is made up of large immunoblastic or plasmablastic cells and anaplastic cells (Jenner et al. 2003). The nuclei in these cells vary from large to round to irregular in shape. Nucleoli are very prominent. The cytoplasm is usually abundant and deeply basophilic. Mitosis is frequent. PEL cells are thought to be derived from post-germinal center B cells (Cesarman et al. 1995). Their immunophenotype is not clear, but they express CD45 and CD138. B- and T-cell markers in the other hand are negative. The immunoglobulin genes of PEL cells are clonally rearranged and hypermutated. PEL cells are known to have about 50 copies per cell of HHV-8 DNA (Cesarman et al. 1995; Asahi-Ozaki et al. 2006) (see also Chap. 18).
20.3.3 Multicentric Castleman Disease
Multicentric Castleman disease (MCD) is a rare polyclonal B-cell angiolymphoproliferative disease characterized by lymphadenopathy, polyclonal hypergammaglobulinemia, and high serum levels of IL-6 (Soulier et al. 1995; Waterston and Bower 2004). MCD affects older patients and has a predilection for men. MCD mainly involves mediastinal lymph nodes but also has been reported in the lung, pleura, and the chest wall (Awotedu et al. 1990; Barrie et al. 1996; Mohamedani and Bennett 1985; Matsuda et al. 1988). Two forms of MCD are recognized: a localized form, also called unicentric, and the multicentric form. Histopathologically, two variants of the disease occur: a vascular hyaline variant (Figs. 20.7 and 20.8) and a plasma cell variant (Figs. 20.9 and 20.10). The vascular hyaline variant is characterized histopathologically by vascular proliferation in germinal centers, while the plasma cell variant, as its name indicates, displays a prominent plasma cell component. HHV-8 has been found to occur in association with multicentric Castleman’s disease (MCD). More than 90 % of AIDS patients with MCD are HHV-8 positive, whereas MCD in the context of absent HIV infection has a HHV-8 prevalence of only 40 % (Parravinci et al. 1997; Katano et al. 2000). HHV-8 can be detected in MCD patients who have polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes (POEMS syndrome) (Bélec et al. 1999). Large B-cell lymphoma may develop in patients with MCD, and with disease progression, frank plasmablastic lymphomas may also develop (Dupin et al. 2000).
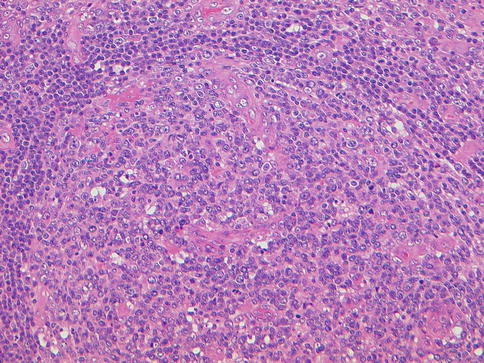
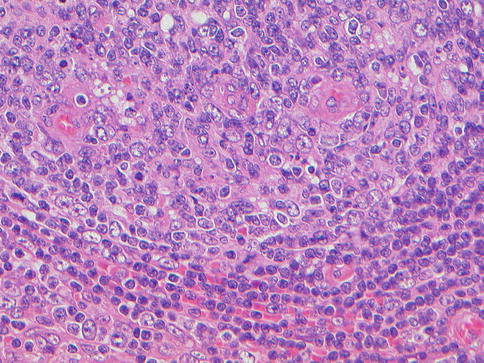
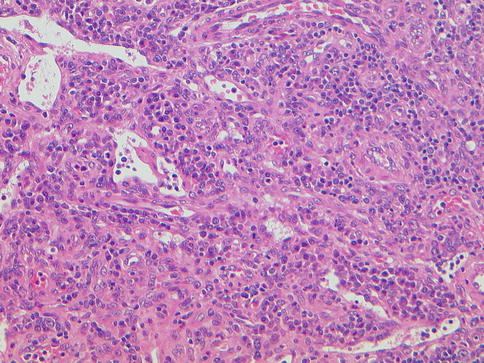
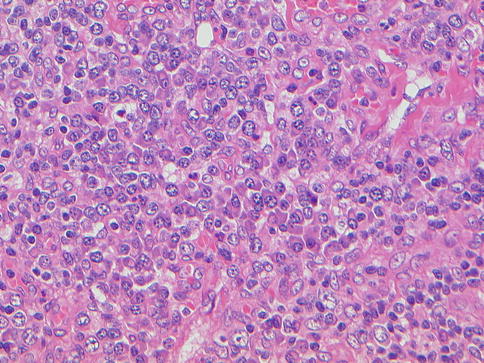
Fig. 20.7
Multicentric Castleman disease, vascular hyaline variant. Note vascular proliferation in germinal centers, low-power view
Fig. 20.8
Multicentric Castleman disease, vascular hyaline variant, high-power view
Fig. 20.9
Multicentric Castleman disease, plasma cell variant. Note marked infiltration by plasma cells, low-power view
Fig. 20.10
Multicentric Castleman disease, plasma cell variant. Note plasma cells with eccentric nuclei, high-power view
20.3.4 Sarcoidosis
Sarcoidosis is a multisystemic granulomatous disease of unknown etiology. Sarcoidosis can involve multiple organs such as the lungs, eyes, heart, lymph nodes, and skin. In the lungs, as elsewhere in the body, the histopathologic hallmark of sarcoidosis is the formation of non-necrotizing granulomas. A report in the mid- to late 1990s described HHV-8 DNA as significantly prevalent in pulmonary tissues, lymph nodes, skin, and oral tissues of 17 patients with sarcoidosis, as compared to tissues from control specimens (Di Alberti et al. 1997). Later, however, another report showed non-detectable HHV-8 DNA in patients with systemic sarcoidosis (Bélec et al. 1998). Thus, there appears to be no consensus on a firm relationship between HHV-8 and sarcoidosis.
20.3.5 Multiple Myeloma
The possible role of HHV-8 in multiple myeloma has been debated ever since a report described an association between the virus and the disease (Rettig et al. 1997). Rettig et al. investigated 15 multiple myeloma patients along with eight patients with monoclonal gammopathy of unknown significance (MGUS). They used PCR to amplify the KS330233 sequence of HHV-8 from bone marrow mononuclear and stromal cells of multiple myeloma patients. Their findings were supported by another study (Said et al. 1997a) showing similar findings in 17 of 20 bone marrow biopsies. Further support for the association was provided by Gao et al. in a report documenting serological evidence of lytic and latent antibodies in two patients with multiple myeloma (Gao et al. 1998). However, a further study by Whitby et al. found latent antibodies in only 4 of 37 patients with multiple myeloma and 2 of 36 MGUS patients, casting doubt on the validity of the association (Whitby et al. 1997).
20.3.6 Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) may be primary (idiopathic), familial, or associated with intrinsic parenchymal lung disease. A characteristic histopathologic feature of primary pulmonary hypertension is the so-called plexiform or angiomatoid vasculopathy. Expression of HHV-8 was first reported in lung tissue from 10 (62 %) of 16 patients with pulmonary arterial hypertension (Cool et al. 2003). Cool et al. found that cells within plexiform lesions as well as cells outside the lesions were positive for latency associated nuclear antigen-1 (LANA-1) by immunohistochemical examination. Tissue from the same 10 patients contained viral cyclin on PCR analysis. Of interest, LANA-1 was not detected in control lung tissue from patients with secondary pulmonary hypertension. The investigators further reported that plexiform lesions from patients with PAH bore histological and immunohistochemical resemblance to cutaneous KS lesions. Further studies (Bull et al. 2003; Nicastri et al. 2005; Gutiérrez et al. 2006) followed supporting Cool’s findings. However, there were also reports (Daibata et al. 2004; Katano et al. 2005; Laney et al. 2005; Henke-Gendo et al. 2005; Galambos et al. 2006; Bresser et al. 2007; Bendayan et al. 2008; Valmary et al. 2011) that offered different results. The relationship of HHV-8 infection and PAH thus remains unclear awaiting further classification. Cool et al. more recently suggested that viral infections alone are unlikely to lead to PAH and that a dysregulated immune system, in conjunction with a viral infection, could be the cause of pulmonary arterial hypertension (Cool et al. 2011).
20.3.7 Inflammatory Myofibroblastic Tumor
The World Health Organization defines “pulmonary inflammatory myofibroblastic tumor” (Yousem et al. 2003) as a histologically variegated set of pulmonary inflammatory pseudotumors, which originally had three subtypes: plasma cell granuloma, fibrous histiocytoma, and organizing pneumonia subtype (Matsubara et al. 1988). Histopathologically, inflammatory myofibroblastic tumor (IMT) is composed of spindle-shaped myofibroblasts, plasma cells, macrophages, and lymphocytes. The clinical behavior of IMT is unpredictable, and cases occurring in children are said to be particularly aggressive (Yousem et al. 2003). Gómez-Román et al. first reported the presence of HHV-8 DNA in seven IMT patients using a combination of nested PCR and Southern blot methods (Gómez-Román et al. 2000). One case of IMT showing HHV-8 antigen in most of xanthomatous macrophages was reported by Farris et al. (Farris and Kradin 2006; Farris et al. 2007). One other report called attention to the detection of HHV-8 in one of 26 cases of IMT (Alaggio et al. 2010). In contrast, the absence of HHV-8 in 20 cases of IMT was reported by Tavora (Tavora et al. 2007).
20.3.8 Interstitial Pneumonias
Interstitial pneumonias, particularly the idiopathic types, are often clinically and radiographically referred to as interstitial pulmonary fibrosis (IPF). Interstitial pneumonias can be idiopathic or secondary to a wide variety of conditions including collagen vascular disorders. For many years, viruses and autoimmunity have been cited as possible etiologic factors. There are several reports calling attention to a possible association of HHV-8 infection with interstitial pneumonias. Trovato et al. at first found HHV-8 in the inflammatory cells within the interstitium endothelial cells of the pulmonary vasculature and in pneumocytes of lung tissue from a patient with chronic interstitial pneumonitis (Trovato et al. 1999). A supporting paper followed, reporting amplification of HHV-8 DNA in lung biopsies from two patients. In situ hybridization and immunohistochemical staining further demonstrated HHV-8-infected lymphoid cells and alveolar macrophages (Müller et al. 2000). Tang et al. studied lung tissue from 23 IPF patients using an organism-specific colorimetric microtiter plate polymerase chain reaction technique and detected one or more herpesviruses in 22 of 23 IPF patients (Tang et al. 2001, 2003). These data added credibility to the concept of an association of HHV-8 infection with interstitial pneumonias. However, negative data was later reported (Zamò et al. 2005).
20.3.9 Other Diseases
There are several reports of other diseases and their possible association with HHV-8, but the available data are sometimes weak or circumstantial, and many have not been confirmed by further investigation in large numbers of patients. For example, HHV-8 infection has been reported in association with multiple sclerosis (Gómez-Román et al. 2000) and congestive heart failure (Ascoli et al. 2002). Serological evidence has also suggested that patients with cardiovascular disease may have a higher prevalence of HHV-8 and HHV-8 DNA as determined in studies based on the analysis of atheromatous plaques (Carletti et al. 2002). Other studies have suggested possible associations of HHV-8 with pemphigus vulgaris (Memar et al. 1997).
20.3.10 Summary
Solid evidence has been presented over the last two to three decades for a strong association between HHV-8 and Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman disease. A host of other associations, as described in this chapter, is less well understood and less well documented. For the moment, the strength of such associations appears to be weak and may be presumed to be incidental and/or circumstantial, at least while awaiting further clarification.
References
Ablashi D, Chatlynne L, Cooper H et al (1999) Seroprevalence of human herpesvirus-8 (HHV-8) in countries of Southeast Asia compared to the USA, the Caribbean and Africa. Br J Cancer 81:893–897PubMedCentralPubMed
Alaggio R, Cecchetto G, Bisogno G et al (2010) Inflammatory myofibroblastic tumors in childhood: a report from the Italian Cooperative Group studies. Cancer 116:216–226PubMed
Armenian HK, Hoover DR, Rubb S et al (1993) Composite risk score for Kaposi’s sarcoma based on a case-control and longitudinal study in the Multicenter AIDS Cohort Study (MACS) population. Am J Epidemiol 138:256–265PubMed
Asahi-Ozaki Y, Sato Y, Kanno T et al (2006) Quantitative analysis of Kaposi sarcoma-associated herpesvirus (KSHV) in KSHV-associated diseases. J Infect Dis 193:773–782, Epub 7 Feb 2006PubMed
Ascoli V, Lo Coco F, Torelli G et al (2002) Human herpesvirus 8-associated primary effusion lymphoma in HIV–patients: a clinicopidemiologic variant resembling classic Kaposi’s sarcoma. Haematologica 87:339–343PubMed
Asher Y, Heller M, Becker Y (1969) Incorporation of lipids into herpes simplex virus particles. J Gen Virol 4:65–76PubMed
Awotedu AA, Otulana BA, Ukoli CO (1990) Giant lymph node hyperplasia of the lung (Castleman disease) associated with recurrent pleural effusion. Thorax 45:775–776PubMed
Barrie JR, English JC, Muller N (1996) Castleman disease of the lung: radiographic, high resolution CT, and pathologic findings. Am J Roentgenol 166:1055–1056
Beksac M, Ma M, Akyerli C et al (2001) Frequent demonstration of human herpesvirus 8 (HHV-8) in bone marrow biopsy samples from Turkish patients with multiple myeloma (MM). Leukemia 15:1268–1273PubMed
Bélec L, Mohamed AS, Lechapt-Zalcman E (1998) Lack of HHV-8 DNA sequences in sarcoid tissues of French patients (letter). Chest 114:948–949PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


