Fig. 9.1
Pressure overload model in the chick embryo. The embryo at day 4 of incubation (left panel) is exposed by amniotomy, and through the developing chest wall, a piece of 10-0 nylon thread is passed under the outflow tract (top right), tied in an overhand knot (middle), and cropped (bottom) to prevent piercing of the fragile surrounding vessels by its sharp ends. Ct conotruncus, FL fore limb bud, RA right atrium, V ventricle
The hemodynamically induced changes in myocardial architecture in this model also could be a substrate of altered electrical pathways. These were investigated as well using optical mapping on isolated hearts [15, 16]. It was found that increased pressure loading accelerated maturation of the ventricular conduction system. At the molecular level, these changes were paralleled by upregulation of conduction system differentiation markers endothelin-converting enzyme 1 (ECE1) and connexin 40 (GJA4).
Another popular model with a long history is left atrial clipping [17, 18] or ligation (LAL [9]) that mimics human hypoplastic left heart syndrome (HLHS). The phenotype of LAL hearts (Fig. 9.2) shows considerable variability from almost normal to extreme involution of the left ventricle with apex-forming right ventricle [19]. Occasional occurrence of VSD in about 25 % of the survivors is similar to results obtained in the left vitelline venous clip model [5] and likewise could be attributed to changes of intracardiac bloodstream patterns [20]. In severe cases, the right atrioventricular valve loses its typical muscular flap-like morphology and resembles a bicuspid fibrous valve. This suggests that hemodynamic stress is an important determinant of morphology of the developing valvular structures and influences differentiation of the connective tissue. In addition, there is a molecular phenotype [21] suggestive of heart failure, and a delay of about 2 days in expression of myocardial differentiation genes (contractile protein isoforms, energetic metabolism enzymes) was detected by microarray analysis in both ventricles. Adaptation of the right ventricle to gradually increasing volume load is progressive. First, there is dilatation with in extreme cases alterations of trabecular orientation from radial to circumferential [9]. Second, there is an increased proliferation within the trabeculae [14] followed by eventual thickening of the compact myocardium, a finding that could be regarded as an acceleration of the normal course of development [22]. These changes in myocardial architecture and proliferative activity could be reversed by subsequent prenatal interventions. Surgical clipping of the right atrial appendage [20] normalized the hemodynamics and rapidly resulted in increased myocyte proliferation in the left ventricle and a tendency toward normalization of reduced left ventricular myocardial volume. This observation demonstrates the feasibility of fetal interventions aimed at mitigating the severity of a number of human congenital cardiac malformations [23, 24].
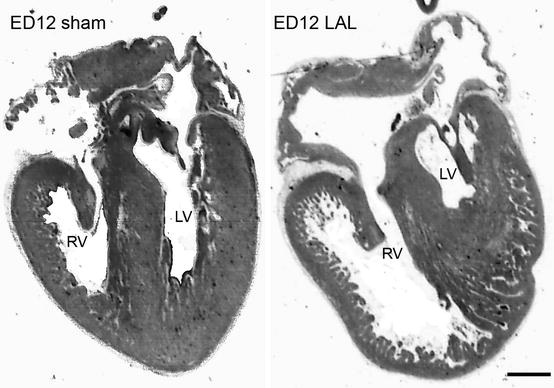
Fig. 9.2
Phenotype mimicking human hypoplastic left heart syndrome induced in the embryonic day 4 chick embryo by partial ligation of the left atrial appendage (LAL). Note the apex-forming right ventricle in the ligated embryo. Scale bar 1 mm
9.4 Prenatal Lamb Model
However simple and self-contained the chick model is, its relevance for human pathology has sometimes been questioned, and fetal mammalian models were thus developed. The lamb is the standard large animal model for investigating intrauterine development of the cardiopulmonary system and testing the effects of various prenatal interventions. In this model it is possible to reproduce typical anatomical and pathophysiological lesions and develop of procedures for their repair. Pioneering surgical studies were performed and summarized by Rudolph [25]. Early investigations demonstrated the suitability of this model for examination of both the pathophysiological consequences of cardiac lesions and the technical methods of intrauterine surgery [26]. With prenatal identification of cardiac lesions, intrauterine cardiothoracic surgery is possible [27, 28]. While a study comparing the effects of early vs. late pulmonary stenosis repair did not show any difference in transverse myocyte diameter even in non-repaired hearts [27], a study comparing the effects of pulmonary stenosis and atresia [28] showed an increase in myocyte width dependent on the level of overload and myocardial weight increase, suggesting a mix of both hypertrophy and hyperplasia. There are thus multiple factors, such as differences in methodological approach, the species used, timing of surgery, and the level of the overload, which influence the nature of myocardial response to prenatal hemodynamic challenge. It appears that even the adult heart, traditionally, and according to the latest evidence, rather incorrectly [29] regarded as a postmitotic organ, is capable of considerable proliferation in response to a long-standing pressure overload [30].
Both the pulmonary artery and aorta were banded in work by McAuliffe and Robbins [31]. This study revealed that only one dominant isoform of cardiac troponin is expressed during fetal and adult development and that expression levels do not change in response to pressure overload. Thus, changes in contractile protein isoform expression (“fetal gene program”) known from adult models of heart overload [32] could not be recapitulated here. This model was used to study the changes in calcium-handling proteins, which could be interpreted as acceleration of the normal course of developmental [33]. At the cellular level, hypertrophy of cardiomyocytes was followed by hyperplasia and decreased proportion of binucleated cells. This is an interesting observation, showing differences from the embryonic chick model as well as the ability of the fetal mammalian heart to behave similarly to the adult heart. As in the case of the embryonic chick pressure overload model, Flanagan and colleagues [34] found that capillary density was maintained in the overloaded group and that the functional flow parameters and coronary resistance were also within the normal range. This shows that the fetal heart adapts its structure in response to pressure challenge (within limits) in a coordinated manner and that the myocardium thus created is normal and well perfused.
Fishman et al. [35] created a prenatal sheep model of the hypoplastic left heart syndrome by obstructing the left ventricular inflow or outflow with a balloon catheter. With the inflow obstruction, within a week, the left to right ventricular weight ratio decreased to 70 % of control, and the mean chamber volume ratio decreased to less than 50 %. The outflow obstruction resulted in less pronounced left ventricular output decrease. Over the long term, the left to right ventricular weight ratio decreased even further, and the left ventricular chamber was nearly obliterated, simulating very severe congenital aortic stenosis. These experiments confirmed the existence of two possible hemodynamic pathways of pathogenesis of hypoplastic left heart syndrome: either through decreased preload or increased afterload of the left ventricle, as seen in fetal mitral or aortic stenosis, respectively.
9.5 Fetal Studies in Rodents
9.5.1 Guinea Pig
An elegant fetal surgical study in small animal models was performed in guinea pigs [36]. Fetal guinea pigs in the third trimester were accessed by hysterotomy, and their ascending aorta was constricted to 50 % of its original diameter. The heart to body weight ratio and left ventricular wall thickness increased significantly in the banded group. There was also a significant increase in percentage of marker of proliferation Ki-67-positive (i.e., proliferating) cells in both ventricles with no changes in apoptosis, suggesting the existence of an adaptation mechanism similar to that observed in the other prenatal models.
9.5.2 Mouse
Mouse embryo is not a frequent subject of hemodynamic studies, mainly because of its small size, the presence of multiple embryos in the same dam making individual longitudinal studies difficult, and general constraints of mammalian model. However, there is wealth of information that could be learned due to availability of transgenic animals mimicking sometimes human cardiac malformations. Basic study of mouse embryonic cardiovascular function and dimensions was performed by Keller et al. [37], replicating similar studies performed previously in chick and rat embryos. Experimental pacing study in murine embryos demonstrated feasibility of Doppler measurements of flows and velocities in this model and confirmed deleterious effects of tachycardia on embryonic cardiac performance [38]. Later studies by this group revealed patterns of response of the embryonic heart rate to hypoxia, which varied according to developmental stage. The initial response to maternal hypoxia was bradycardia, followed at reoxygenation by either overshoot prior to return to baseline (paralleling the maternal response), continued bradycardia, or simple return to normal rate [39]. Pharmacological study of effects of moderate doses of caffeine [40] showed a slight but significant deleterious effect on embryonic growth and cardiac performance, possibly mediated via adenosine receptor blockade.
Phoon and colleagues investigated with ultrasound biomicroscopy mice with NFATc1 deletion [41]. These mice lack the outflow valves and die rather suddenly at mid-gestation. It was shown that the decline is indeed rapid, with bradycardia preceding the embryonic demise while the myocardial contractile function remained preserved despite regurgitant blood flow propagating back to the placental circulation.
9.5.3 Rat
Rat embryos were also used as models for pharmacological testing (recently reviewed by Sedmera et al. [42] from the perspective of fetal arrhythmias). Of course, significant arrhythmias often result in hemodynamic instability and could lead to fetal complications. Another recent report from rat model was investigating the pathogenesis of preeclampsia induced by inflammatory reaction after lipopolysaccharide injection [43].
9.6 Fetal Hemodynamics in Humans
The ultimate goal of studies performed in various animal models (Fig. 9.3) is to gain better understanding into working of prenatal human circulation, especially at stages which are not easily studied in depth (particularly the first trimester).
As mentioned above, the fetal hemodynamics is dependent upon normal and regular heart rate [42]. Alterations in hemodynamics are believed to be one of the causes for cardiac malformations such as hypoplastic left heart syndrome [19] or valvar atresias (Fig. 9.2). Abnormalities in cardiac performance can bear influence on all the systems in the developing fetus, including the central nervous system [44]. Experimental interventions aimed at normalizing hemodynamics and preventing irreversible changes include balloon aortic or pulmonary valvuloplasty [24, 45]. However, a recent American Heart Association statement [46] lists these treatment options as experimental, as not all of them might translate into desirable outcome despite technical success [47]. This highlights the need of further basic as well as translational research into mechanisms of interactions between the structure and function of the developing heart.
9.7 Postnatal Hemodynamics
Abrupt changes in circulation occurring after birth have been the subject of numerous studies, and there is considerable variation among species in, e.g., mechanisms of closure of the foramen ovale (cell proliferation closing multiple foramina in birds or flap-like valve sealing the double septum in mammals). The neonatal heart temporarily retains some of the characteristics of the fetal heart, notably the proliferative activity that is used to compensate altered functional demands [48]. As we have recently reviewed this issue in depth [33], we may refer the readers to this book.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


