Fig. 13.1
(a) Immunofluorescence for CXCL12, CXCR4, CXCR7, and c-kit in mice lung exposed to chronic hypoxia (10 % oxygen for 3 weeks) or normoxia. Scale bars = 50 μm; (b) Pulmonary perivascular c-kit+ cell infiltration in mice exposed to chronic hypoxia (10 % oxygen for 3 weeks) treated or not with AMD3100 (a CXCR4 antagonist; intraperitoneal injection: 10 mg/kg/day) and CCX771 (a CXCR7 antagonist; intraperitoneal injection: 10 mg/kg/day) or both. #: p < 0.0001. Reproduced with permission of the European Respiratory Society. Eur Respir J 2011 37:1392–1399; published ahead of print 2010, doi:10.1183/09031936.00045710
13.3 Chronic Hypoxia-Induced Pulmonary Vascular Remodelling
13.3.1 The Physiological Adaptive Response to Hypoxia
The pulmonary circulation is unique among vascular beds, characterized by a high flow, low-resistance, low-pressure system. Although all of the cardiac output is pumped through the pulmonary circulation, the measured values of mean pulmonary arterial pressure (mPAP) in pulmonary arteries for a resting, healthy adult human is about 14 ± 3 mm of mercury (Hg), when systemic arterial blood pressure is about 100 ± 20 mmHg. This unique feature of the pulmonary circulation is mainly explained by two important characteristics of the pulmonary vasculature: a high compliance of pulmonary pre-capillary arterioles characterized by a thin vessel wall and a high capacity to recruit vessels available to accommodate increase in flow. Indeed, the system can accommodate flow rates ranging from about 6 L/min under resting conditions to 25 L/min in strenuous exercise, with minimal increases in pulmonary pressures. In contrast to systemic arteries, the pulmonary artery carries oxygen-poor blood and the pulmonary vein carries oxygen-rich blood. In addition, oxygen deprivation causes vasoconstriction of the small pulmonary arteries, when systemic arteries dilate under hypoxic conditions. This phenomenon, known as hypoxic pulmonary vasoconstriction (HPV), is more pronounced as the vessel diameter decreases and represents an important physiological mechanism by which pulmonary arteries constrict in hypoxic lung areas in order to redirect blood flow to areas with greater oxygen supply.
Prolonged hypoxia, however, is a critical pathological stimulus that affects the expression/activity of the major regulators of vascular function necessary for maintenance of homeostasis in both pulmonary and systemic circulations. Such regulators include production of vasoconstrictors and vasodilators, activators and inhibitors of smooth muscle cell growth and migration, pro-thrombotic and anti-thrombotic mediators, as well as pro- and anti-inflammatory signals. Prolonged exposure to a hypoxic environment leads to increased pulmonary vascular resistance due to progressive structural and functional adaptations of the pulmonary vascular bed. There are age and/or sex differences and these pulmonary vascular remodelling can be variable between individuals, probably based on genetics and adaptive mechanisms. The magnitude of these responses is intense in pig, horse, and cow, moderate in rodent and human, and very low in dog, guinea pig, yak, and lama (Grover et al. 2011). Importantly, chronic hypoxia-induced lesions, such as high altitude pulmonary edema (HAPE) in humans or chronic hypoxia-induced PH in animal models, are reversible. Interestingly, HAPE is mostly dependent on individual susceptibility as the acute and excessive rise of mPAP (35–55 mmHg) within few days after arrival at high altitude, which precedes the formation of pulmonary edema, occurs in less than 5 % of the mountaineers and climbers. Recent elegant studies demonstrated, in cattle susceptible to brisket disease, that this response is inherited, though the identification of the genetic basis is the subject of ongoing research (Newman et al. 2011). Beside individual susceptibility, the extent of the structural and functional changes of the vascular bed in humans depends on the type, the severity, and the duration of hypoxemia. The predominant cause of PH development is represented by alveolar hypoxia due to chronic lung diseases, impaired control of breathing, or residence at high altitude as reported above. Among chronic lung diseases, the diseases most commonly associated with PH, which is usually moderate, are chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), and a syndrome combining pulmonary fibrosis and emphysema (CPFE). The prevalence of PH varies depending on the associated disease and its severity. Clinical and epidemiological data highlight the notion that only a small percentage of patients with chronic lung diseases develop severe PH (defined as mPAP above 35–40 mmHg), clearly suggesting the need of additional “hit(s)” for the development of chronic hypoxia-induced severe PH.
13.3.2 Mechanisms Underlying Chronic Hypoxia-Induced Pulmonary Vascular Remodelling
The chronic hypoxia-induced remodelling of the pulmonary circulation takes place sequentially and includes: (a) endothelial dysfunction with release of vasoactive mediators, growth factors, cytokines, and chemokines; (b) smooth muscle cell hyperplasia and hypertrophy with resultant pulmonary arterial medial wall thickening; (c) adventitial fibroblast proliferation, accumulation of extracellular matrix components, and myofibroblast differentiation (Stenmark et al. 2000, 2002); and (d) changes in pulmonary pericyte behaviour including proliferation, migration, and differentiation (Ricard et al. 2014). These alterations in structure and function of the lung vasculature are accompanied by pronounced changes in cell metabolism, gene expression, and cytoskeletal organization. During hypoxic exposure, cells switch from oxidative metabolism to anaerobic glycolysis for energy production to sustain the same level of cell function. In addition, it is now well-recognized that prolonged hypoxia results in an inflammatory phenotype in the lung, both in humans and in animals (Carpenter and Stenmark 2001; Frid et al. 2006; Frohlich et al. 2013; Kubo et al. 1996; Madjdpour et al. 2003; Marsch et al. 2013; Minamino et al. 2001; Schoene et al. 1988; Vergadi et al. 2011; Zampetaki et al. 2003; West et al. 2004). Elevated levels of pro-inflammatory cytokines (i.e., IL- 1β, IL-6, IL-8, and TNF-α) and increased numbers of neutrophils and macrophages have been detected in bronchoalveolar lavage fluid of humans exposed to hypobaric hypoxia (Kubo et al. 1996; Schoene et al. 1988; West et al. 2004). These observations may explain the positive effects of corticosteroids prophylaxis on acute hypoxic hypertension in adults prone to HAPE (Fischler et al. 2009; Maggiorini et al. 2006). Finally, there is clear evidence that moderate hypoxia stimulates angiogenesis and increases vascular permeability in the pulmonary circulation, potentially explained by alterations of pulmonary pericyte functions or vessel coverage (Ricard et al. 2014). These phenomena may potentially contribute to the formation of alveolar edema in acutely hypoxic lung (Howell et al. 2003; Hyvelin et al. 2005).
The molecular networks underlying chronic hypoxia-induced pulmonary vascular remodelling are not fully understood; however, integrative genomic analyses have revealed a panel of key regulated genes and transcriptional factors that may contribute to hypoxia-induced phenotypic changes. HIF-1α and HIF-2α are among these master regulators of hypoxic signalling (Schofield and Ratcliffe 2004). Lowered oxygen tension inhibits hydroxylation of proline and asparagine residues by prolyl hydroxylase domain-containing proteins (PHDs) and stabilizes or induces a rapid nuclear translocation of cytoplasmic HIF-1α and/or HIF-2α that heterodimerizes with HIF-1β (also known as ARNT) and subsequently leads to induction of gene transcription and adaptive responses that accommodate cellular function in ischemia or hypoxia. In addition to HIF activation, hypoxia has been demonstrated to activate several other transcription factors including to varying degrees: Egr-1 (Bae et al. 1999; Jin et al. 2000; Nishi et al. 2002; Yan et al. 1998, 1999), nuclear factor-kB (NF-kB) (Chandel et al. 2000b; Koong et al. 1994; Leeper-Woodford and Detmer 1999; Liu et al. 2014; Matsui et al. 1999; Schmedtje et al. 1997; Scholz and Taylor 2013; Taylor et al. 2000), cyclic AMP response element-binding protein (CREB) (Beitner-Johnson and Millhorn 1998; Comerford et al. 2003), activating protein (AP)-1 (Laderoute 2005; Minet et al. 2001; Salnikow et al. 2002:53; Alarcon et al. 1999; Chen et al. 2003; Sermeus and Michiels 2011), Sp1, Sp3 (Deacon et al. 2012; Discher et al. 1998; Yang et al. 2014), and CCAAT/enhancer-binding protein β (C/EBPβ) (Semenza 2000; Yan et al. 1995; Matsui et al. 1999; Park and Park 2010; Larsen et al. 2008). In addition, reactive oxygen species (ROS) (Chandel et al. 1998, 2000a, b; Chandel and Schumacker 2000; Killilea et al. 2000; Zepeda et al. 2013), calcium (Pamenter and Haddad 2014; Peers and Kemp 2004; Zepeda et al. 2013), and intracellular cyclic AMP levels (Peers and Kemp 2004) or the response to extracellular adenosine have been demonstrated to participate in the modulation of the molecular networks underlying chronic hypoxia-induced pulmonary vascular remodelling. Low oxygen availability also controls mRNA concentrations by inducing changes in microRNA transcription and expression (Nallamshetty et al. 2013). All of these mediators lead to a cellular response when this balance between oxygen supply and demand is disturbed.
Acute (normobaric 10 % FiO2 for 10–30 min) and chronic hypoxia (normobaric/ hypobaric 10 % FiO2 for 3–6 weeks) are among the most widely used models to study pulmonary vasoreactivity and PH, respectively (Ryan et al. 2011; Stenmark et al. 2009; Weir and Archer 1995). They are very valuable to test new targets and/or treatments and to give insights into the disease mechanisms. Animal PH models allow study of the early phases of disease process, at several different levels of cell function and also help to analyse precise mechanisms of action of potential agents for clinical use. However, chronic hypoxia-induced pulmonary vascular lesions are reversible when back to normoxic conditions, clearly demonstrating that such animal models do not reproduce the full spectrum of changes seen in lung specimens from severe PH patients with advanced pulmonary arterial hypertension (PAH). Therefore, efforts have been made to improve this chronic hypoxia-induced PH by treating rats with a single dose of VEGF-receptor antagonist (SU5416) plus hypoxia (de Raaf et al. 2014; Ryan et al. 2011; Stenmark et al. 2009; Taraseviciene-Stewart et al. 2001). However, pulmonary vascular lesions experimentally induced, even in this latter animal model, do not mimic human disease in particular regarding the plexogenic lesions. Furthermore, a low mortality rate is often reported, contrasting with the devastating prognosis of human PAH. In addition, interspecies-, age-, sex-related differences have been reported.
13.4 HSCs and Chronic Hypoxia-Induced Pulmonary Vascular Remodelling: Is There Any Link?
Our group has reported that idiopathic PAH patients display high serum CXCL12 levels and that pulmonary endothelium represents one local abnormal source (Montani et al. 2011) (Fig. 13.2). We have also demonstrated that remodelled vessels from chronically hypoxic mice exhibit a marked induction of CXCL12 and its receptors, CXCR4 and CXCR7 (Gambaryan et al. 2011) (Fig. 13.1). In the chronic hypoxia-induced PH mouse model, we found that administration of AMD3100, a CXCR4 antagonist, alone or combined with CCX771, a CXCR7 antagonist, prevented vascular remodelling, PH, and perivascular accumulation of c-kit+/sca-1+ progenitor cells, with a synergistic effect of these agents (Gambaryan et al. 2011) (Fig. 13.1). CXCL12 represents an important factor for recruitment and mobilization of progenitor cells, in particular HSCs expressing the transmembrane tyrosine kinase receptor c-kit (although c-kit may be present on other cell types such as mast cells). Interestingly, c-kit+ cells can be mobilized from the bone marrow and may potentially differentiate into vascular cells to promote myocardial repair after infarction (Orlic et al. 2001) and to contribute to atherosclerosis (Sata et al. 2002). Furthermore, it is known that c-kit+ HSCs can be mobilized from the bone marrow into the systemic circulation by hypoxia (Parmar et al. 2007; Takubo et al. 2010; Imanirad and Dzierzak 2013; Lord and Murphy 1973; Murphy and Lord 1973; Serebrovskaya et al. 2011). Interestingly, an increase in c-kit+ cell number was found in remodelled pulmonary arterial wall in chronically hypoxic mice (Davie et al. 2004; Gambaryan et al. 2011) and in idiopathic PAH (Montani et al. 2011) (Figs. 13.1 and 13.3). Moreover, a work from Launay and colleagues shows a role of 5-HT2B receptors in the maturation of different bone marrow-derived cells and in their localization in the pulmonary vascular wall in the chronic hypoxia-induced PH mouse model (Launay et al. 2012).
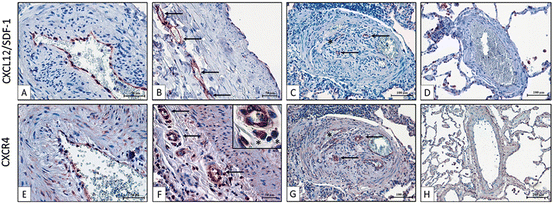
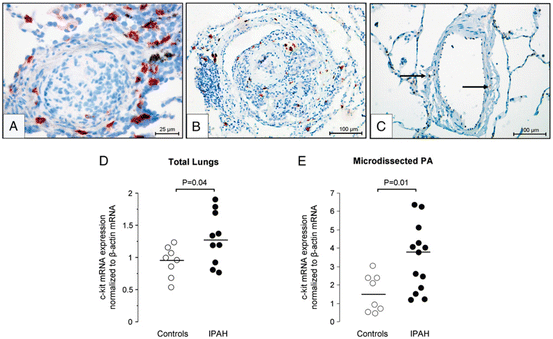
Fig. 13.2
Immunochemistry of CXCL12 and CXCR4 in pulmonary arterial lesions from patients with idiopathic pulmonary arterial hypertension (IPAH) (a–c and e–g) and controls (d, h). In panels b and f, arrows indicate the vasa vasorum and asterisks indicate inflammatory cells. In panels c and g, arrows and asterisks indicate endothelial cells. Reprinted with permission of the American Thoracic Society. Copyright © 2014 American Thoracic Society. Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, Huertas A, Hammad H, Lambrecht B, Simonneau G, Launay JM, Cohen–Kaminsky S, Humbert M (2011) C–kit–positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. American journal of respiratory and critical care medicine 184 (1):116–123. doi:10.1164/rccm.201006–0905OC. Official Journal of the American Thoracic Society
Fig. 13.3
Immunohistochemistry for c-kit (red staining) in paraffin-embedded lung sections: from patients with idiopathic pulmonary arterial hypertension (IPAH) (a, b) and controls (c). Real-time polymerase chain reaction quantifications of c-kit mRNA levels normalized to β-actin in total lungs from controls and IPAH patients (d) and in microdissected small pulmonary arteries (PA) from controls and IPAH patients (e). Reprinted with permission of the American Thoracic Society. Copyright © 2014 American Thoracic Society. Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, Huertas A, Hammad H, Lambrecht B, Simonneau G, Launay JM, Cohen-Kaminsky S, Humbert M (2011) C–kit–positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. American journal of respiratory and critical care medicine 184 (1):116–123. doi:10.1164/rccm.201006–0905OC. Official Journal of the American Thoracic Society
By highlighting the CXCL12 signalling pathway dysregulation and the increased numbers of c-kit+ cells in human PAH and experimental PH, we raise the question of the role of HSCs in pulmonary vascular remodelling and disease progression: whether HSCs can be recruited and mobilized in the lung and whether these cells can differentiate into vascular cells and actively participate to lesion formation and pulmonary vascular remodelling needs to be addressed.
Unfortunately, there is no robust data on the role of HSCs in chronic hypoxia-induced pulmonary vascular remodelling. The available evidences so far have been obtained using bone marrow cells without clear consensus about HSC markers, cell phenotypes, disease read-outs, and animal tools. In the first study, Hayashida and colleagues used mice transplanted with bone marrow from enhanced green fluorescence protein (GFP)-transgenic mice. After 8 weeks, they exposed the transplanted mice to chronic hypoxia, showing GFP+ cells in pulmonary arterial wall, including into the adventitia. In addition, most of GFP+ cells expressed α-smooth muscle cell actin, suggesting that these cells can differentiate in situ (Hayashida et al. 2005). In the second study, Raoul and coworkers showed no effect of intravenous administration of bone marrow cells on pulmonary hemodynamic measurements (right ventricular systolic pressure), on right ventricular hypertrophy, and on the percentage of muscularized vessels, suggesting that HSCs had no role in the disease development and/or progression (Raoul et al. 2007). Based on these observations, it is therefore difficult to conclude whether HSCs contribute to pulmonary vascular remodelling and, if so, by which underlying mechanisms. Additional studies are clearly needed to define and understand the potential role of HSCs in chronic hypoxia-induced pulmonary vascular remodelling.
13.5 Concluding Remarks
In this chapter, we have reviewed the current knowledge on the role of HSCs in chronic hypoxia-induced pulmonary vascular remodelling. Unfortunately, we still lack a clear understanding of HSC behaviour, activation, recruitment, and potential differentiation in the pulmonary vasculature, particularly under chronic hypoxia. A consensus on techniques such as bone marrow transplantation, animal models, and tools to follow HSCs in a time-dependent manner in the setting of chronic hypoxia would help defining whether these cells are mobilized in a chronic hypoxia-dependent manner, whether they are recruited in the pulmonary vasculature, and whether they are able to differentiate into pulmonary wall cells and contribute to and/or delay the progression of pulmonary vascular remodelling.
References
Abraham M, Biyder K, Begin M, Wald H, Weiss ID, Galun E, Nagler A, Peled A (2007) Enhanced unique pattern of hematopoietic cell mobilization induced by the CXCR4 antagonist 4F-benzoyl-TN14003. Stem Cells 25(9):2158–2166. doi:10.1634/stemcells.2007-0161 CrossRefPubMed
Alarcon R, Koumenis C, Geyer RK, Maki CG, Giaccia AJ (1999) Hypoxia induces p53 accumulation through MDM2 down-regulation and inhibition of E6-mediated degradation. Cancer Res 59(24):6046–6051PubMed
Bae SK, Bae MH, Ahn MY, Son MJ, Lee YM, Bae MK, Lee OH, Park BC, Kim KW (1999) Egr-1 mediates transcriptional activation of IGF-II gene in response to hypoxia. Cancer Res 59(23):5989–5994PubMed
Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, Calandra G, Bridger G, Dale DC, Srour EF (2005) Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med 201(8):1307–1318. doi:10.1084/jem.20041385 CrossRefPubMedCentralPubMed
Carpenter TC, Stenmark KR (2001) Hypoxia decreases lung neprilysin expression and increases pulmonary vascular leak. Am J Physiol Lung Cell Mol Physiol 281(4):L941–L948PubMed
Chandel NS, Schumacker PT (2000) Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol 88(5):1880–1889PubMed
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95(20):11715–11720CrossRefPubMedCentralPubMed
Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT (2000a) Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J Biol Chem 275(33):25130–25138. doi:10.1074/jbc.M001914200 CrossRefPubMed
Chandel NS, Trzyna WC, McClintock DS, Schumacker PT (2000b) Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J Immunol 165(2):1013–1021CrossRefPubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


