15 Hematolymphoid Disorders
In the interval since the first edition of this textbook, there have been clinically significant revisions to the classification of lymphomas that affect the approach to diagnosing hematolymphoid proliferations. These include the identification of new “phenotypic” entities, such as the anaplastic large B cell lymphoma associated with ALK translocations,1–5 plasmablastic microlymphoma,6,7 cyclin D1+ large B cell lymphomas,8 and new “cytogenetic” entities, such as “double-hit” diffuse large B cell lymphomas.9 Other changes include the subdivision of old entities, such as diffuse large B cell lymphoma into genetic “activated” and “germinal center” subtypes.10–12 Diagnostic categories, such as atypical Burkitt lymphoma and Hodgkin-like anaplastic large T cell lymphoma, have been retired. This is evident in the revisions of the World Health Organization (WHO) publication on the pathology and genetics of hematopoietic and lymphoid tissues,5 which increasingly represents a clinicopathologic touchstone for oncology.
Twenty-first-century care requires both the generalist and the specialist to be familiar with the indications for the full range of molecular tests available, the specimen requirements for each, and their suitability and relevance to specific differential diagnostic setting. When presented with hematologic proliferations in the lung, pathologists must use an expanding array of special studies to formulate and resolve the differential diagnosis and provide the results of tests that aid in risk stratification.11 Microarray technology is an established method for both class discovery and class definition13,14 and in the not-too-distant future may become a part of the diagnostic and prognostic workup of patients with lymphoma.15 This chapter describes the tests and techniques of hematopathology and the diagnoses that these tests enable.
Special Studies
Immunohistochemistry
Specimen Requirements
Immunophenotypic analysis of tissue sections is an essential part of the evaluation of hematolymphoid proliferations in both nodal and extranodal sites. The range of antibodies and the increase in the number of rabbit monoclonal antibodies available for paraffin-embedded material continues to expand,16–19 but most differential challenges can be resolved with a handful of markers. Table 15-1 lists markers that cover all of the entities described in this chapter. Boxes 15-1 and 15-2 describe panels and the manner in which they might be used.
Table 15-1 Antibodies Useful in the Paraffin Section Evaluation of Hematolymphoid Proliferations
| Marker | Description |
|---|---|
| B Cell Lineage Markers | |
| CD10 | Positive in follicle center cell non-Hodgkin lymphoma (not lineage-specific; also present on some epithelial and stromal tumors) |
| CD19 | Early B cell marker (also present on B-LBL; not present on plasma cells) |
| CD20 | Mature B cell marker (not present on B-LBL; most plasma cells negative) |
| CD23 | Activated B cells |
| CD79a | Immature and mature B cells (present on B-LBL as well as plasma cells) |
| PAX5 | Immature B cells, including lymphoblasts, mature B cells; negative in plasma cells; also positive in some neuroendocrine tumors, including small cell carcinoma |
| CD138 | Plasma cells, some cases of classic Hodgkin lymphoma |
| MUM1 | In the proper context, postfollicular B cells and plasma cells |
| IgD | Immunoglobulin heavy-chain delta, present in benign mantle cells, some lymphomas |
| κ, λ | Immunoglobulin light chains (cell surface expression assessed by flow; cytoplasmic expression by immunohistochemistry) |
| T Cell Lineage Markers | |
| CD1a | Some immature T cells (thymocytes), Langerhans cells |
| CD2 | Pan T cell marker; may also be present on natural killer cells by flow cytometry |
| CD3 | T cells |
| CD4 | Helper/suppressor T cells |
| CD5 | Preferential T cell marker (also positive in some B cell neoplasms) |
| CD7 | T cells, some natural killer cells |
| CD8 | Cytotoxic T cells |
| CD43 | Preferential T cell marker (also positive in some B cell neoplasms and granulocytic proliferations) |
| CD56 | Natural killer cells and some T cells; also positive in some neuroendocrine tumors |
| CD57 | Natural killer cells and some T cells; also expressed on some neuroendocrine tumors |
| Monocyte/Macrophage/Accessory Cell Markers | |
| CD1a | Langerhans cells, some T cells (thymocytes) |
| CD14 | Monocytes (paraffin markers available, not widely used) |
| CD15 | Granulocytes, also positive in Hodgkin lymphoma and adenocarcinoma |
| CD21 | Follicular dendritic cells, some B cells |
| CD31 | PECAM-1; marks vascular endothelium and monocytes, macrophages, and histiocytes |
| CD33 | Granulocytes (paraffin marker available, not yet widely used) |
| CD68 | Macrophages, monocytes (two clones; KP1 and PGM1 have slightly different specificities) |
| CD163 | Hemoglobin scavenger receptor; expressed on macrophages and histiocytes, including histiocytic malignancies |
| Langerin | Langerhans cells, both in Langerhans cell histiocytosis and Langerhans cell sarcoma |
| Miscellaneous Markers | |
| ALK1 | Positive in some peripheral T cell lymphomas, also in some inflammatory myofibroblastic tumors |
| bcl6 | Transcriptional regulator positive in germinal center B cells as well as some lymphoblasts; may be positive in the lesional cells of some T cell neoplasms |
| bcl2 | Anti-apoptosis protein positive in virtually all lymphoid proliferations except benign germinal center B cells and Burkitt lymphoma |
| Cyclin D1 | Cell cycle regulator positive in mantle cell lymphoma, myeloma, and rare cases of large B cell lymphoma |
| CD45 | Leukocyte common antigen present on lymphocytes, blasts, monocytes, and L&H type Reed-Sternberg cells |
| Oct2 | Transcription factor in some B and T cells; also present in L&H type Reed-Sternberg cells |
| TdT | Terminal deoxynucleotidyl transferase, a marker of the blastic stage |
| EMA | Epithelial membrane antigen (positive in some large cell lymphomas and some plasmacytomas) |
| Ki67 | Proliferation marker that helps to identify proliferation centers in chronic lymphocytic lymphoma/small lymphocytic leukemia and is also useful in the multiparameter distinction of high-grade large B cell lymphoma and Burkitt lymphoma |
B-LBL, B cell lymphoblastic lymphoma: L&H, lymphocytic and histiocytic; PECAM, platelet-endothelial cell adhesion molecule.
Box 15-1 Immunophenotypic Profiles Associated with Lymphoid Neoplasia
Phenotypic Findings Indicative of Lymphoid Neoplasia
Aberrant Gain of a Preferential T Cell Marker on a Proliferation of B Cells
Phenotypic Findings Helpful in Classifying Neoplastic Small Lymphoid Proliferations
MUM1 Expression
Box 15-2 A Panel Approach to Immunophenotypic Analysis of Hematolymphoid Proliferations in the Lung
The judicious and cost-effective use of immunostains is maximized if the pathologist puts each marker ordered to specific purpose.20–23 In the workup of hematolymphoid lesions of the lung, there are four principal goals:
In working up hematologic tumors in the lung, the pathologist needs to be aware of certain pitfalls.20 It has long been recognized that CD138 (syndecan 1) marks both plasma cells and a range of epithelial tumors. More recently, PAX-5 has been shown in a broad range of neuroendocrine tumors,31 including small cell carcinoma and Merkel cell carcinoma.32 These studies show that, especially in the evaluation of lung tumors, no one marker should be the sole basis on which the B cell nature of a lung process is defined.33 Other pitfalls were reviewed recently by Yaziji and Barry.20
Flow Cytometry
Specimen Requirements
An advantage of flow cytometry over tissue section immunophenotyping is that three or four markers can be evaluated simultaneously on specific small- and large-cell populations. Through this detailed multiparameter profile, many lymphoproliferative disorders can be classified more accurately.34–37 However, flow cytometric phenotyping does not allow for visualization of the immunoarchitecture of the proliferation, which can be an important element of accurate classification of lymphomas, particularly in the lung, where marginal zone lymphoma (MaZL) is so common. In tissue section-based immunophenotyping, the pathologist can readily identify situations in which the lesional foci have disappeared from the sections used for the stains. In flow cytometric immunophenotyping, however, the pathologist must ensure that the lesional cells are present on a cytospin made from the disaggregated cells.
Most reference laboratories performing flow cytometry have established panels for specific clinical scenarios (lymphoma panel, adult leukemia panel, pediatric leukemia panel, expanded T cell panel) that are tailored to efficiently identify the classic immunophenotypic profiles characteristic of common disease entities.38–41 The appropriate panel should be used to address the diagnostic question. Perhaps 10% to 15% of the time, however, a particular lymphoma or leukemia will have a variant phenotype.41,42 Examples include a lack of CD10 in a lymphoma of follicular origin, acquisition of CD10 in hairy cell leukemia, CD5 expression in large cell lymphoma, CD19 expression in acute myeloid leukemia, apparent dim CD23 expression in mantle cell lymphoma, and a lack of CD23 expression in chronic lymphocytic leukemia. For this reason and because the immunoarchitecture is an important part of the disease definition for some lymphomas, both flow cytometry and immunohistochemistry may need to be performed in some cases, and the diagnostician should not be shy about doing both.
Cytogenetics
Specimen Requirements
Many leukemias and lymphomas have recurring chromosomal translocations that can be identified with FISH and classic cytogenetic testing.43–53 These tests are widely available at many referral laboratories, but they are expensive and time-consuming. Therefore, the pathologist must be aware of their indications, applications, and limitations and also must make the clinician aware of the time frame between biopsy and final results.
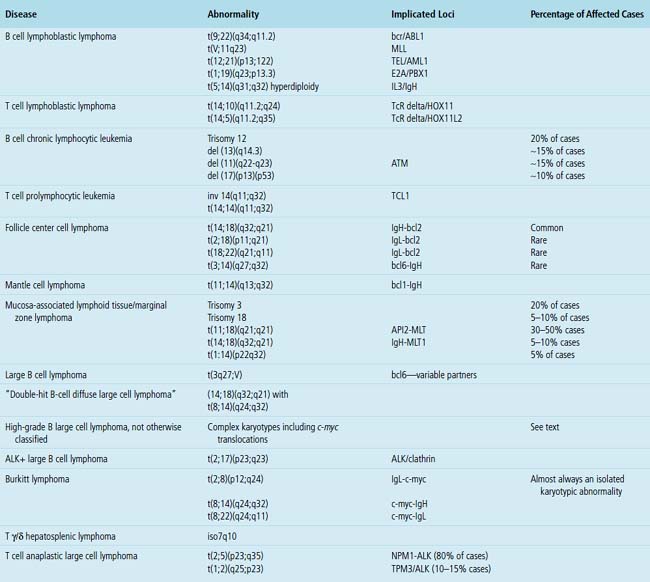
One of the most important applications of cytogenetics is in distinguishing between morphologically similar tumors with widely different aggressiveness or treatment protocols. The distinction between small lymphocytic lymphoma and mantle cell lymphoma is an example of the first type, and the distinction between Burkitt lymphoma and high-grade B cell lymphoma is an example of the second type. Although some translocations are characteristic of certain diseases, not all are as specific for a particular entity as once believed. For instance, c-myc–related translocations were once believed to be specific for Burkitt lymphoma, but are now known to be present in some large B cell lymphomas and in some lymphomas that populate the new WHO category of “B cell lymphoma unclassifiable with features intermediate between high-grade large B cell lymphoma and Burkitt lymphoma”5,48 (discussed later). The most common and clinically relevant karyotypic changes related to leukemias and lymphomas are enumerated in Table 15-2.
Molecular Genetics
Specimen Requirements
Over the last decade, molecular genetic analysis has gone from being an expensive test seldom ordered by non-hematopathologists to one routinely ordered when an atypical lymphoid infiltrate is encountered and flow cytometry was not performed or the results were not informative.36 General pathologists must be familiar with the range of molecular tests available, their applications and limits, and the small but real risk of misleading results. Although B5, Bouin, and Hollande fixatives yield excellent cytologic detail, they also denature DNA to the point that tissues fixed in these solutions are not usable for molecular analysis.54–57 Therefore, care should be taken during prosection to include sufficient tissue in formalin to allow for molecular studies if preliminary findings on touch preparations or frozen section suggest an hematolymphoid process.58
Polymerase chain reaction analysis may be used on formalin-fixed, paraffin-embedded material or archived snap-frozen tissue to document clonality,59–62 define lineage, evaluate for translocation events, and assess for the presence of infectious agents. PCR may also be used to speciate mycobacterial organisms. Clonality is assessed by examination of areas of the immunoglobulin heavy- and light-chain loci or the T cell receptor α/β or γ loci that are rearranged during normal lymphoid development.54 Specially designed primer sets that flank certain gene loci are used for PCR-based evaluations for certain translocations, a helpful solution when there are no commercially available FISH probes or if tissue suitable for FISH is not available.
As “objective” as they are, molecular genetic tests do not replace the thought process of diagnosis. These test results are adjunctive data that point in one direction or another, but the final diagnosis must be made by the pathologist.63 The results of molecular studies must be integrated into the “big picture” painted by the clinical setting, the morphologic findings, and the immunophenotypic results. Although clonality is used to define neoplasia, lack of clonality does not prove that a lesion is reactive. The finding of an oligoclonal band is meaningless to the treating physician unless the pathologist puts the result into the context of the morphologic and clinical data. Low tumor cell numbers in Hodgkin lymphoma, lymphomatoid granulomatosis, and T cell–rich B cell lymphoma may yield nonclonal results because of the dilutional effect of reactive cells. Similarly, non–B, non–T cell malignancies, such as natural killer cell lymphomas and myeloid leukemias, yield a polyclonal smear because the lesional cells are of neither B cell nor T cell lineage.
Normal Lymphoid Tissue in the Lung and the Concept of MALT
The lung contains an extensive lymphatic network that channels antigen-rich lymph centripetally toward the parenchymal, septal, hilar, and mediastinal lymph nodes. Organized lymphoid tissue in the periphery of the normal lung is limited to sparse submucosal aggregates of lymphocytes and intrapulmonary lymph nodes, but can be more substantial centrally and along bronchiolar branch points.64 Inhaled particulate matter is trapped in the mucus layer of the proximal airways, and some passes across patches of specialized epithelium, where it initiates primary and secondary immune responses. Inhaled irritants stimulate a principally monocyte/macrophage response, whereas inhaled immunogens promote a lymphocytic (or lymphoplasmacytic) response. In practice, inhalational exposures are seldom purely one or the other, and so the tissue response tends to be mixed and is not infrequently masked by fibrinous exudates and actively phagocytic macrophages. The lymphoid tissue proliferates as a result of nonexogenous stimuli as well. In autoimmune conditions and immunodeficiency states, there is an intrinsic dysregulation of lymphoid proliferation, and the lymphoid hyperplasia—“acquired mucosa-associated lymphoid tissue” (MALT) or “bronchus-associated lymphoid tissue”36,65–67 is less masked by acute-phase mucosal changes. Intrapulmonary lymph nodes, also a part of the pulmonary immune surveillance system, are uncommon, but when found, they are more often solitary, peripherally located, and located in the lower lung field. Their structure and immunoarchitectural compartments—and the diseases that affect them—are no different from those of extrapulmonary lymph nodes.
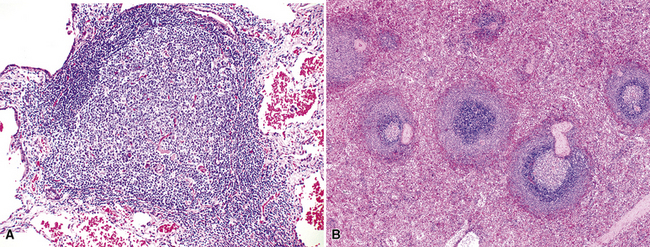
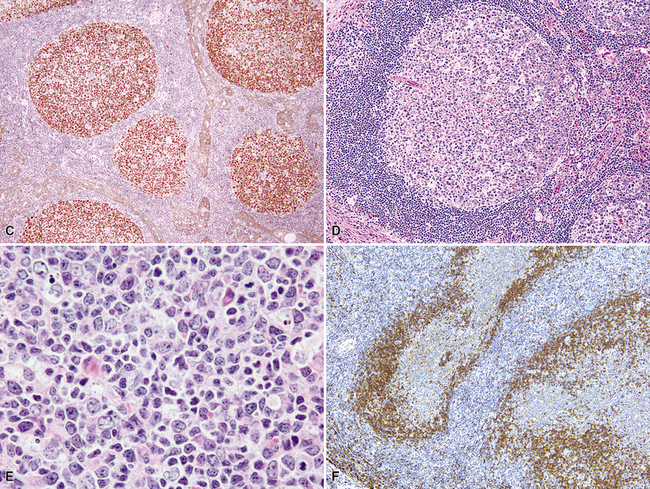
In addition, MALT has a close and specific relationship to the adjacent alveolar and bronchiolar epithelium, the “lymphoepithelium,” where antigen processing and presentation occur. Benign MALT has a distinctive immunoarchitecture, with discrete compartments: the B cell–rich follicles, the mantle, and the T cell–rich interfollicular regions. In some cases and in some areas, a marginal zone of intermediate-sized cells with moderate to abundant amounts of cytoplasm may be interposed between the mantle and the interfollicular regions, although this marginal zone is never as well developed as it is where MALT was originally recognized, in the spleen and Peyer patches (Fig. 15-1). The lymphocytes in these structures shuttle between the mucosa and the circulation to provide ongoing immune surveillance and response to antigens that diffuse through the airways. Between follicles, lymphocytes range from small and resting to intermediate in size and somewhat activated. Plasma cells may be commingled, none with Dutcher bodies. Where immunoblasts are present, their regular size, round nuclear shape, and smooth nuclear contour support a benign interpretation.
Very limited foci of lymphocytes likely have no clinical or radiologic correlate and may not require recognition with a diagnostic line in the report. However, lymphoid accumulations that either have a radiologic correlate or clearly associate with distortions of airways or air spaces should be mentioned (Box 15-3 and Table 15-3).
Box 15-3 Hyperplasia of Mucosa-Associated Lymphoid Tissue: General Features
What Should Be Present
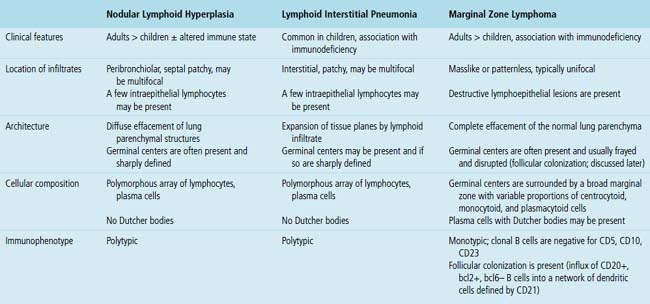
Reactive Lymphoid Proliferations
Clinicopathologic Patterns of Pulmonary Lymphoid Hyperplasia
The three main patterns of lymphoid hyperplasia—follicular bronchiolitis (FB), nodular lymphoid hyperplasia (NLH), and diffuse lymphoid hyperplasia (lymphoid interstitial pneumonia [LIP])—may be seen in isolation or may coexist in the same specimen.67,68 Much of the work in identifying specific benign and malignant lymphoid proliferations requires the diagnostician to recognize the intactness or the patterned disruption of the morphologic and immunologic landmarks of normal MALT (Fig. 15-2; see also Box 15-3 and Table 15-3).
Follicular Bronchiolitis
Follicular bronchiolitis is slightly more common in males than in females, involves the lungs bilaterally, and produces a centrilobular reticulonodular pattern of involvement on radiologic studies.69,70 In exceptional cases, opacities up to 1 cm in size may be seen. FB is most commonly seen in patients with congenital or acquired immunodeficiency (HIV, common variable immunodeficiency, or immunoglobulin A deficiency), collagen vascular disease (especially rheumatoid arthritis), or chronic obstructive pulmonary disease,70–73 and it may also be seen at the periphery of localized infectious processes of the lung.
Microscopically, the key feature is multiple foci of eccentric peribronchiolar accumulations of lymphoid tissue that distort and may narrow the bronchiolar lumen71,74–76 (Box 15-4 and Fig. 15-3). Confluent nodule-forming infiltrates larger than 1 cm should raise concern about lymphoma. The structure of benign MALT is preserved, with bcl2– germinal centers that are crisply demarcated by an immunoglobulin D–positive mantle zone and a polymorphous lymphocytic and histiocytic component at the interface with normal lung parenchyma. The proliferation may compress the airways, leading to postobstructive bronchiectasis in distal parenchyma. There is no interstitial involvement in the alveolar walls away from the bronchioles, and the air spaces are uninvolved (Fig. 15-4), a feature that distinguishes FB from LIP.69,70,75–77
Box 15-4 Features of Follicular Bronchiolitis
What Should Be Present
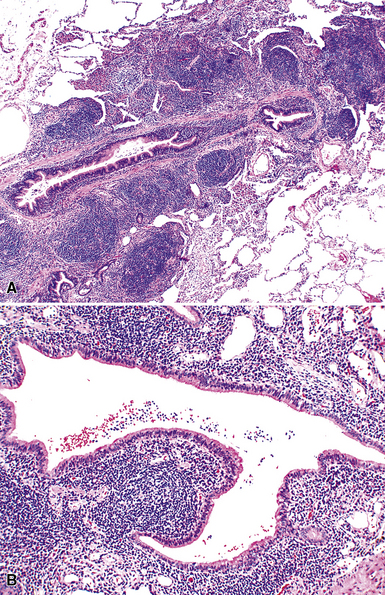
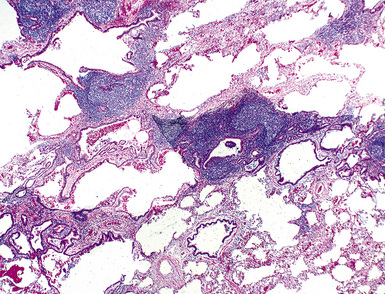
Differential diagnostic considerations include nonspecific chronic inflammation,78,79 which is not organized or airway-centered, and which usually extends into alveolar walls. NLH may enter into the differential diagnosis, and the distinction is as much quantitative as it is a perception of a mass-forming process that compresses adjacent normal lung parenchyma.80Constrictive bronchiolitis may be associated with lymphocytic accumulations around the bronchioles, but the cue to the correct diagnosis is concomitant peribronchiolar fibrosis with reduction in lumen size such that the bronchiole is significantly smaller in diameter than its accompanying arteriole. An elastin stain may be helpful in defining the architecture in such circumstances.
Nodular Lymphoid Hyperplasia
Nodular lymphoid hyperplasia is an extremely rare condition, with only one large series published, representing the Armed Forces Institute of Pathology experience over a decade. In older literature, “NLH” has been used synonymously with “pseudolymphoma,” an outdated and confusing term that should be discarded.80 It is a proliferation that is most commonly seen in adults, reported in the second to ninth decade. Whereas there are reports of NLH in patients with an altered immune state, such as autoimmune disorders, collagen vascular disease, or acquired immunodeficiency, in the Armed Forces Institute of Pathology series there was no special relationship with those conditions.80–82
Patients come to clinical attention because of cough or reasons unrelated to respiratory symptoms. One or several discrete subpleural or peripheral nodules are detected on radiography.81 If there is a reticulonodular pattern in the remaining lung, clinical concern for lymphoma as well as infectious etiologies is greater. In contrast to the lymphoid lesions of FB, the nodules of NLH are typically larger than 0.5 cm, but seldom larger than 5 cm.
The lymphoid infiltrate of NLH forms a circumscribed nodule of lymphoid tissue (Box 15-5 and Fig. 15-5), with intact architecture, including germinal centers, a discrete mantle, and a preserved “paracortical” interfollicular zone. The process is not pleurally based, and there should not be a bronchiolar distribution.
Box 15-5 Features of Nodular Lymphoid Hyperplasia
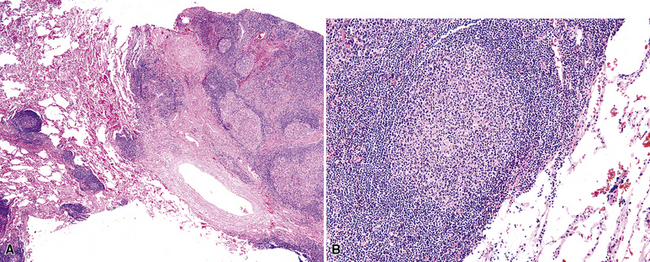
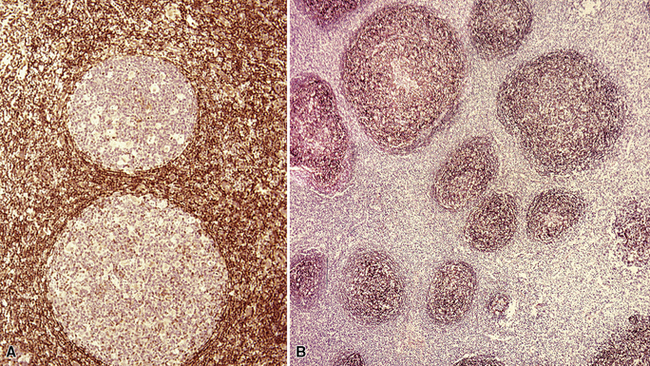
Figure 15-5 Nodular lymphoid hyperplasia is a discrete mass-forming lymphoid proliferation that is spherical or ovoid, in contrast to the linear parabronchial distribution of follicular bronchiolitis. A, Germinal centers are widely spaced, vary in size, and maintain the benign immunoarchitectural landmarks seen in Figure 15-2. B, Pale-staining monocytoid cells seen in marginal zone lymphoma are not present.
Within the mass, follicular architecture predominates.80,83 Cells within nodules retain centrocytic and centroblastic morphologic features, and the mantle zone retains its population of small cells with deeply basophilic round nuclei and scant cytoplasm. Small resting lymphocytes, stromal elements, and plasma cells fill the interfollicular zone, which may also contain patchy accumulations of histiocytes. Lesional foci of NLH remain sharply circumscribed from the surrounding lung parenchyma, with at least a partial immunoglobulin D–positive mantle (Fig. 15-6), without significant extension along the lobar septa or into the alveolar walls, in contrast to LIP. Plasma cells with Russell bodies and Mott cells may be present, but destructive lymphoepithelial lesions, Dutcher bodies, and follicular colonization84 should not be identified.
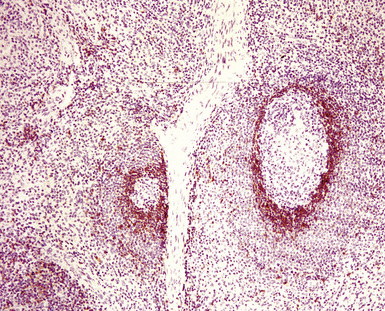
If the nodule was not sampled for flow cytometry, a useful immunohistochemical panel includes CD20, CD21, CD3, bcl2, bcl6, and immunoglobulin D, as well as kappa, lambda, MUM1, and cytokeratin. The immunoglobulin D–positive mantle should be present and fairly crisply demarcated from the immunoglobulin D–negative germinal center inside and the immunoglobulin D–negative paracortex beyond. The germinal center cells should have a bcl2–, bcl6+ phenotype. The bcl6+, CD20+ B lymphocytes within the follicles are definitionally polytypic, as are the plasma cells and immunoglobulin D–positive mantle cells. The interfollicular areas are rich in CD3+ T cells. CD21 staining in NLH highlights the compact nature of the follicular dendritic cell network (Fig. 15-7). A disrupted appearance, particularly if associated with a significant bcl2+, bcl6–, or MUM1+ population of B cells within the follicles or an increase in the number of interfollicular B cells, suggests the diagnosis of MaZL (see Table 15-3).82
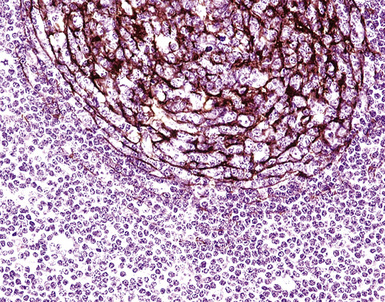
Figure 15-7 The CD21+ follicular dendritic meshwork of nodular lymphoid hyperplasia is sharply circumbscribed, in contrast to the ragged appearance of the network in marginal zone lymphoma, which is seen in Figure 15-17B.
Because of its rarity, NLH is a diagnosis that should be approached with caution and made only after all necessary studies to exclude lymphoma are performed. In practice, if neither flow cytometry nor immunohistochemistry yields a secure diagnosis, molecular studies are a reasonable final step. Principal microscopic differential considerations in transbronchial or transthoracic biopsy specimens may include a particularly robust FB, LIP, and various low-grade lymphomas. In contrast to FB, NLH is mass-forming and displaces significant amounts of lung parenchyma.80LIP is readily excluded by radiologic correlation because it is generally diffuse and bilateral and is not mass-forming, and it is primarily interstitial, with extensive infiltration of the alveolar walls, whereas NLH displaces normal lung tissue as a mass.
In contrast to follicular lymphoma, the germinal centers of NLH are widely spaced, vary in size, exhibit light zone/dark zone polarity, and are demarcated by a distinct immunoglobulin D–positive mantle zone composed of cytologically bland small lymphocytes.5 The finding of bcl2 positivity within nodules in excess of what CD3+ intrafollicular T cells would yield, or the finding of significant numbers of bcl6+, CD20+ B cells outside of the germinal centers should raise concern about follicular lymphoma. If flow cytometry is not available, molecular assessment for clonality and disease-defining translocations should be pursued (PCR, FISH). MUM1 immunostaining may be helpful in distinguishing follicular lymphoma from MaZL.
Marginal zone lymphoma is the most challenging element of the differential diagnosis. Because MaZL is much more common than NLH, it can be argued that molecular studies should be pursued in all cases before a benign diagnosis is rendered. The architecture may be identical to that of NLH, or there may be some blurring of the mantle zone separating the germinal centers from the intervening cellularity. A significant population of monocytoid cells (intermediate size, round or reniform nucleus, sufficient quantities of pale cytoplasm that nuclei are widely spaced on routine sections), either within or between nodules, favors MaZL. The nodules may exhibit “follicular colonization” by MaZL,84 which is seen as displacement of the CD20+, bcl6+, bcl2–, MUM1– centrocytes and centroblasts of the normal follicle by the MaZL cells (CD20+, bcl6–, bcl2+, MUM1+; discussed later). The mantle zone in MaZL is eroded or distorted on immunoglobulin D stain, and the follicular dendritic cell meshwork is diffused through the dilutional effects of the infiltrating MaZL cells. Although they are not diagnostic of MaZL, destructive lymphoepithelial lesions should be sought on cytokeratin stain.
Lymphoid Interstitial Pneumonia and Diffuse Lymphoid Hyperplasia
The pattern of LIP may be seen in both children and adults, and up to 40% of cases are eventually attributable to a specific underlying condition. It is more common in the setting of altered immune states, such as autoimmune conditions and connective tissue disorders (especially Sjögren syndrome85), AIDS,86,87 and congenital immunodeficiency states,88 and after bone marrow transplant. It may also be a tissue response pattern to infections such as mycoplasma, chlamydia, Epstein-Barr virus (EBV), and legionella.89 In some series, females are affected disproportionately,69 perhaps because of the common collagen vascular disease association. Older literature doubtless includes cases of MALT lymphoma, which may skew both outcome and the clinicopathologic parameters that have been associated with LIP.
Patients with LIP present with a cough and slow but progressive shortness of breath, and radiologic studies usually show bilateral basilar patchy opacities or reticulonodular infiltrates.69,87,89–91 Cysts have been reported in LIP,92 but correlation with clinical findings suggests that the cysts are more likely a manifestation of the underlying condition (Sjögren syndrome) than of LIP. Some patients have systemic symptoms, such as fever and weight loss, and many (70%) have polyclonal hypergammaglobulinemia; both the clinical and laboratory abnormalities likely reflect the underlying altered immune state.
In contrast to FB and NLH, the infiltrate of LIP has a dominant interstitial pattern of distribution, although the constituent cellular components are otherwise similar. Small, cytologically bland lymphocytes and intermingled plasma cells distend the alveolar walls, with accentuation along the bronchovascular bundles and lobular septae (Box 15-6 and Fig. 15-8). Aggregates of histiocytes or poorly formed granulomas may be present, but neutrophils and eosinophils are scarce. A few germinal centers may be present.69,87,90 An intraepithelial component mimicking the lymphoepithelial lesions of MALT lymphoma and a coalescence in and around the microvasculature have been described, but truly destructive changes are not part of LIP (Fig. 15-9).
Box 15-6 Features of Diffuse Lymphoid Hyperplasia/Lymphoid Interstitial Pneumonia
What Should Be Present
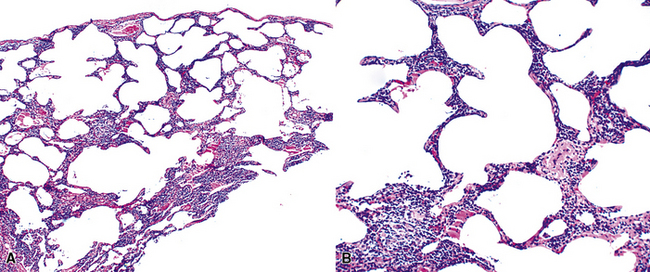
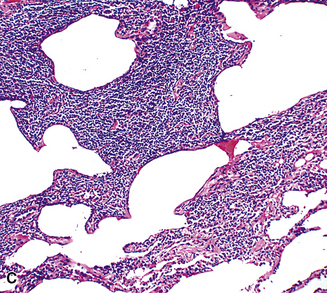
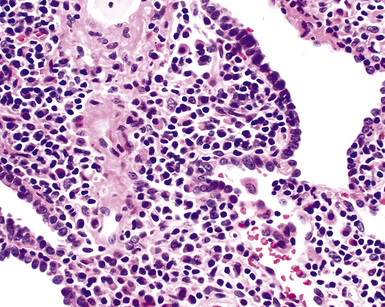
Figure 15-9 Lymphoid interstitial pneumonia shows composition by morphologically mature lymphocytes. Most are CD3+ T cells, a helpful feature in distinguishing this condition from marginal zone lymphoma. Lymphocytes may abut the epithelium of the distal airways, but they do not form destructive aggregations, as seen in marginal zone lymphoma and in Figure 15-15.
An LIP-like pattern has been described in the lung in the setting of atypical infectious mononucleosis.53,93,94 In these few reports, the alveolar walls and interstitium were expanded by a mixture of lymphocytes, plasma cells, and transformed lymphocytes (immunoblasts), and there was a patchy alveolar exudate. In situ hybridization for EBV-encoded ribonucleotides (EBERs) is the most sensitive and specific means of identifying the virally mediated nature of the process. Immunosuppressed patients are at increased risk for EBV-related lymphoid proliferations, and biopsy is usually undertaken to assess for a specific infectious process. In the transplant setting, the terminology of post-transplant lymphoproliferative disorders (PTLDs) should be used (discussed later).
Occasional germinal centers and focal intraepithelial accumulations of lymphocytes may prompt consideration of MaZL/MALT lymphoma, but the bilateral and interstitial (rather than unifocal and mass-forming) nature of LIP, as well as the lack of a dominant B cell population in the interfollicular areas, provides a strong and objective means of excluding this possibility. Because of the interstitial distribution, dominant T cell population, and cytologic heterogeneity of LIP, distinction from pulmonary presentation of systemic lymphomas, such as small lymphocytic lymphoma, mantle cell lymphoma, or follicle center cell lymphoma is seldom an issue. In difficult cases, or when diagnostic material is limited, immunohistochemistry (CD20, CD3) usually permits a definite diagnosis (discussed later; see Box 15-3 and Table 15-3).
Castleman Disease
Castleman disease includes two distinct conditions. One, the “hyaline vascular variant,” is almost invariably presents via mass effect of a solitary mediastinal mass or central lymphadenopathy in otherwise asymptomatic individuals and is cured by complete resection. The other, the “plasma cell variant,” often manifests with systemic symptoms and multifocal disease, and has significant associated morbidity and mortality. What brings them together95 is that, first, the two histologies may coexist in the same lymph node, and second, they were characterized in separate publications by the same individual, Benjamin Castleman.
Hyaline Vascular Variant
The hyaline vascular variant of Castleman disease (HVCD) arises in axial node groups of the mediastinum and abdomen, although it may extend along the hilum to involve peribronchial lymph nodes. Most patients have no systemic symptoms and present because of mass effect (e.g., airway compression or superior vena cava syndrome) or have mediastinal adenopathy detected during radiologic studies performed for other reasons.96–98 A patient who has “B” symptoms and diffuse adenopathy likely has a mixed type of Castleman disease, in which the histologic features of the plasma cell variant are present elsewhere (discussed later). In contrast to the plasma cell variant of Castleman disease (PCCD), there is no consistent abnormality in laboratory findings.
At low power, the architecture is nodular, composed of small and involuted germinal centers with expansive immunoglobulin D–positive mantles formed of small lymphocytes, often in a laminated or “onion skinning” array. Multiple germinal centers may be found in the boundaries of a single mantle zone, and in opportune sections, the lymphoid depletion in the germinal centers unmasks the radially penetrating high endothelium—the “lollipop” motif (Box 15-7 and Fig. 15-10). Sinuses between the regressed follicles are imperceptible usually because they are absent. At high power, the germinal centers are depleted of lymphocytes and contain both extracellular matrix and abundant follicle dendritic cells. The interfollicular zone includes plasmacytoid monocytes, stromal myoid cells, histiocytes, dendritic cells, and lymphocytes.99–101
Box 15-7 Features of Castleman Disease, Hyaline Vascular Type
What Should Be Present



