Hematologic Effects and Coagulopathy
Andreas Pape
Kai Zacharowski
Circulating blood is the transport medium of oxygen, nutrients, carbon dioxide, and cellular and soluble factors of the immune and coagulation systems (i.e., white blood cells, immunoglobulins, platelets, and coagulation factors). Hematorheologic properties including blood viscosity and viscoelasticity are essential for the transport of these substrates, in particular at the site of the microcirculation. Additionally, a sophisticated balance between pro- and anticoagulant factors maintains blood fluidity, on the one hand, while providing hemostatic potential to seal off a microvascular bleeding at any time, on the other hand.
Cardiopulmonary bypass (CPB) imposes extremes on hematorheology and on hemostasis, for example, by hemodilution, hypothermia, hemolysis, and heparinization. Moreover, the CPB surface is perceived as a foreign body by circulating blood elements, which attempt to “clot it off” by activating hemostasis and simultaneously to “reject it” by mounting an inflammatory attack. As a consequence, coagulation gets activated, resulting in consumptive coagulopathy and platelet dysfunction.
Both proinflammatory mediators and activated clotting factors enter into the patient’s circulation, thereby promoting mechanisms of organ dysfunction. In addition, the impairment of hemostasis results in increased bleeding and transfusion requirements, which additionally increase perioperative morbidity and mortality.
This chapter will present the pathophysiologic principles of hematologic and coagulatory disorders provoked by CPB on cellular and soluble components and the cross talk between these single factors.
CELLULAR COMPONENTS
Red Blood Cells
Red blood cells (RBCs) represent about 99% of cellular blood constituents and are therefore the major determinant of hematocrit (Hct). Hct, defined as the percentage of packed cell volume in whole blood, is the key determinant of oxygen transport capacity, on the one hand, and of blood viscosity and thus hemorheologic properties, on the other hand. During CPB, oxygen transport and hemorheology become impaired by hemodilution, hypothermia, and hemolysis.
Priming the CPB circuit with acellular fluids results in a dilution of the cell mass within the vascular system, that is, in acute anemia with a corresponding decrease of hematocrit and oxygen transport capacity. Depending on the type of the CPB circuit and the patient’s circulating blood volume, the degree of hemodilution ranges between 25% and 35% (1) (for more details, see Chapter 16). Physiologically, acute anemia is compensated by increases in cardiac output and arteriovenous oxygen extraction (2). However, during CPB, the possibility to increase cardiac output is limited, so that increasing oxygen extraction becomes the predominant compensatory mechanism in this setting. Thus, a sustained decrease of mixed venous oxygen saturation might indicate a critical limitation of oxygen transport capacity (3).
Hemodilution exerts effects on coagulation beyond the dilution of coagulatory factors, since RBCs contribute to hemostasis: first, RBCs are integrated into the red thrombus like bricks, thereby contributing to clot stability in a passive manner; second, RBCs expose pro-coagulatory factors like phosphatidylserine on their surface, which directly contribute to thrombin generation (4); third, the laminar blood flow of RBCs directs platelets and coagulation toward the endothelium (“radial dispersion”) where they are needed for hemostasis (5). Due to different shear rates within the arterial and the venous circulations (see below), the latter effect plays a predominant role in arterial blood vessels.
To decrease total body oxygen demand, CPB is usually performed under hypothermia (see Chapter 9). The consequence is a left shift of the oxygen dissociation curve with impaired oxygen offloading from hemoglobin to the tissues. Moreover, blood temperature influences blood viscosity. It has long been known that rheologic properties of blood are impaired by increased blood viscosity during hypothermia (6), so that hypothermia alters both oxygen transport and hematorheologic properties.
Another frequently observed consequence of CPB is hemolysis, which is principally caused by mechanical shear stress and turbulences within the roller pump and the circuit (i.e., tubes, connectors, cannula, reservoirs, and oxygenator) (7). Damaging mechanisms include the contact of circulating blood with air and nonendothelial surfaces, wall impact
forces, pressure gradients to assist venous drainage, and the use of cardiotomy suction (8).
forces, pressure gradients to assist venous drainage, and the use of cardiotomy suction (8).
While some RBCs disintegrate immediately, a considerable quantity of erythrocytes suffer “sublethal” damage, resulting in cellular lysis at a later time point. However, the deformability of these RBCs is altered, leading to an impaired blood rheology. As a result of disintegration of erythrocytes, hemoglobin and heme molecules are released into the circulation. The degree of hemolysis seems to correlate with the duration of CPB (8) and is reflected by increased plasma levels of free hemoglobin and lactate dehydrogenase (LDH) and by decreased levels of haptoglobin. As RBCs suffering a “sublethal” damage may break down at a later time point, the release of intracellular constituents into the circulation can be sustained over a long period of time and free hemoglobin levels may even increase after the cessation of CPB (9).
Free hemoglobin is a potent scavenger of nitric oxide (NO), the most important endogenous vasodilator. Reduced bioavailability of NO exerts vasoconstriction, which has proven harmful for microcirculatory function and thus tissue oxygenation (10). Moreover, free heme reacts with endogenous hydrogen peroxide, resulting in the formation of radical oxygen species and thus in the induction of oxidative damage (11).
A direct consequence of vascular dysfunction is an impairment of organ perfusion, so that the role of hemolysis in the development of organ dysfunction after surgery with CPB has gained increasing interest (8). To decrease morbidity and mortality, therapeutic targets address the modification of CPB circuit systems and scavengers of free hemoglobin molecules (12,13).
Circulating blood behaves like a non-Newtonian fluid, as the viscosity of flowing blood varies with shear rates. Aside from hematocrit, blood viscosity is determined by RBC deformability, RBC aggregation, and plasma viscosity (14). Within an arterial blood vessel, laminar blood flow can be modeled as concentric rings of fluid moving at different velocities (see Fig. 17.1). The central core of fluid moves at the highest velocity. Sheathing this central, fastest-moving fluid core is a ring whose velocity is slightly slower. Immediately outside that ring is another that moves still more slowly, as does each successive ring of blood all the way out to the vessel wall. The shear rate, γ, is a measure of how rapidly these adjacent fluid layers slide past each other, and is expressed in inverse seconds (s-1)10. The shear rate at a radial point, r (measured from the vessel center), can be calculated according to the following equation:
γ = dvz/dr = 4(vz) r/R2
where vz is the mass average velocity and R is the vessel radius. Accordingly, the shear rate is greatest at the vessel wall (γwall = 4(vz)/R) and zero at the center of the vessel. Given that wall shear rates are inversely proportional to the vessel radius, R, wall shear is greatest in arterioles, in the order of 5,000 s-1, and decreases in progressively larger arteries, reaching a still-forceful nadir of approximately 500 s-1 in the major arteries. Contrasting this, the velocity of blood flow, vz, is low enough in the venous system that shear force is virtually negligible in the venous circulation (15).
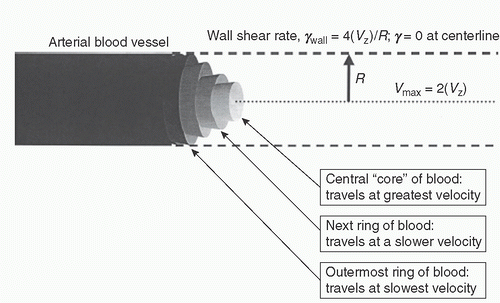 FIGURE 17.1. Rheology of fluid flow through a cylinder. Schematic of fluid flowing as concentric rings of a Newtonian fluid moving at different velocities within a cylinder. The velocity is greatest at the center and decreases with radial distance from the center, with VZ = the mass average velocity. The shear is greatest at the wall and approaches zero at the center. (Adapted from Rinder CS. Hematologic effects and coagulopathy. In: Gravlee GP, Davis RF, Stammers AH, et al., eds. Cardiopulmonary bypass—principles and practice. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2008:439–458.) |
The differing shear rates within the arterial and venous circulation also affect the coagulant effects within these different vascular regions. In the “pressurized” arterial system, a relatively small vascular disruption can rapidly result in significant blood loss and hematoma formation, creating the need for a system that can speedily and securely seal off
bleeding sites. Platelets dominate this “rapid response team,” initially containing blood loss, then providing a surface on which to localize and accelerate the fibrin formation that ultimately consolidates hemostasis. In the venous circulation, by contrast, the more leisurely flow rates diminish the need for speed, making platelets less critical, and indeed, thrombin generation constitutes the pivotal reaction controlling the balance of venous hemostasis. It is critical to understand how venous and arterial hemostasis operate in part through common pathways, and how the two differ, to fully appreciate the manifestations of different CPB-induced coagulation defects.
bleeding sites. Platelets dominate this “rapid response team,” initially containing blood loss, then providing a surface on which to localize and accelerate the fibrin formation that ultimately consolidates hemostasis. In the venous circulation, by contrast, the more leisurely flow rates diminish the need for speed, making platelets less critical, and indeed, thrombin generation constitutes the pivotal reaction controlling the balance of venous hemostasis. It is critical to understand how venous and arterial hemostasis operate in part through common pathways, and how the two differ, to fully appreciate the manifestations of different CPB-induced coagulation defects.
White Blood Cells
Contrasting RBCs, white blood cells (WBCs) make up approximately 1% of whole blood volume under physiologic conditions. As cells of the immune system, WBCs serve the purpose to defend the organism against infections and foreign materials. Basically, WBCs can be divided into granulocytes (including neutrophils, basophils, and eosinophils) and agranulocytes (lymphocytes, monocytes, and macrophages).
The contact of circulating blood with the nonendothelial surface of the CPB circuit initiates a marked activation of circulating WBCs—predominantly neutrophils and monocytes—resulting in a proinflammatory response. In detail, mechanisms of CPB-associated inflammation are described in Chapter 13.
During CPB, WBCs get activated by mediators of the contact and complement systems (predominantly by kallikrein and C5a), by elevated plasma levels of heparin, histamine, thrombin, and FXIIa, and by proinflammatory cytokines (16,17). C5a induces neutrophil chemotaxis, superoxide generation, and degranulation (18).
Activated neutrophils release cytotoxic enzymes (neutrophil elastase, cathepsin G, lysozymes, myeloperoxidase) triggering the formation of oxygen free radicals and hydrogen peroxide. Moreover, activated neutrophils interact with endothelial cells upon expression of their adhesion factor (CD11b/CD18), thereby promoting leukocyte rolling and sticking, clumping, microvascular occlusion, and finally end-organ ischemia (19). Following transmigration into extravascular tissues, cytotoxic effects of activated neutrophils and the formation of tissue edema result in the deterioration of cellular function (20,21,22).
Compared with neutrophils, the activation of monocytes during CPB happens more slowly, as peak levels of monocyte activity have been observed with a certain delay (19). The mechanism leading to the activation of monocytes during CPB has not been fully elucidated. However, the alternative pathway of complement activation may be involved, but interactions with soluble tissue factor and endotoxin are additionally discussed (23).
Activated monocytes release proinflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-8 (24), thereby reinforcing the inflammatory response to CPB. By expression of tissue factor, activated monocytes also promote the generation of thrombin and thus the activation of the coagulation system (25). Contrasting this, a newly discussed anti-inflammatory effect of monocytes is mediated by the CD163 receptor. Elevated levels of CD163 expression were observed on activated monocytes after endocytosis of hemoglobin-haptoglobin complexes, which elicits a marked release of anti-inflammatory IL-10 (26).
Activated leukocytes contribute significantly to hemostatic disorders during CPB (16), in particular by expressing tissue factor on their surfaces (27), thereby activating factor VII and initiating the plasmatic coagulation system (see below). Additionally, activated monocytes have been reported to conjugate with platelets via the granule membrane protein-140 receptor, thereby contributing to thrombopenia and coagulopathy (28,29).
Anticoagulant responses mediated by leukocytes include the release of elastase and cathepsin G as well as the upregulation of the adhesion receptors CD11b/CD18, the latter facilitating the adhesion to fibrin (30). Additionally, lysosomal enzymes such as elastase and cathepsin G are capable of degrading fibrin (31,32), thereby contributing to the destabilization of clots.
Platelets
Structure and Function of Platelets
Platelets are anucleate cell fragments derived from megakaryocytes of the bone marrow. Within the circulation, the life span of platelets is approximately 7 to 10 days. The abundance ratio of platelets to RBCs is 1:10 to 1:20. Resting platelets are biconvex discoid structures measuring 2 to 3 µm in diameter. The cellular membrane possesses invaginations known as the open canalicular system, which permits small molecules to enter into the network of internal membranes. Inside the cells are two types of granules, the α- and dense granules, which store molecular platelet activators (15). The more prevalent a-granules mainly contain adhesive ligands and growth factors (e.g., GPIIb/IIIa, fibrinogen, von Willebrand factor [vWF]), while dense granules contain calcium, serotonin, and adenosine diphosphate (ADP) (33).
In the case of bleeding, platelets initiate primary hemostasis by adhesion to the endothelial defect. Initial platelet adhesion is driven by interactions between the GPIbα receptor and subendothelial compounds like vWF and collagen, which is exposed due to the disruption of the endothelial cell layer.
Essential to the adhesion of platelets to the endothelium is vWF, a large multimeric glycoprotein (20,000 kDa) composed of base units (polypeptides of 2,813 amino acids) which are produced by endothelial cells, megakaryocytes, and in the subendothelial tissue (34). Primarily, platelet adhesion to the endothelium is supported by two domains of the vWF multimer: the α1 domain, which binds to the platelet GPIbα receptor and the α3 domain binding to subendothelial collagen.
In the plasma, vWF circulates as a loosely coiled molecule with an apparent diameter of 200 to 300 nm. As the α1
domain remains cryptic in that state, circulating vWF usually shows no affinity for cocirculating platelets. However, under the influence of the high shear present along the vessel wall (a threshold value of 1,000 s-1 is required), vWF uncoils to lengths as great as 1,300 nm and the α1 domain becomes apparent, thereby enabling the interaction with the platelet surface receptor GPIbα (34).
domain remains cryptic in that state, circulating vWF usually shows no affinity for cocirculating platelets. However, under the influence of the high shear present along the vessel wall (a threshold value of 1,000 s-1 is required), vWF uncoils to lengths as great as 1,300 nm and the α1 domain becomes apparent, thereby enabling the interaction with the platelet surface receptor GPIbα (34).
The coupling of this vWF domain to GPIbα is characterized by a high rate of bond formation. This “fast-on” binding tethers the platelet to the exposed subendothelium in the face of the high-velocity blood flow that would otherwise sweep the platelet past the bleeding site. The relatively weak strength of this adhesive tethering is soon overcome by the local shear force, and the platelet moves on, albeit now traveling at a slower velocity but still in proximity to the vessel wall (15). In addition to slowing the platelet’s velocity, this brief vWF-GPIbα interaction stimulates platelet activation with a conformational change in the GPIIb/IIIa receptor, which allows it to interact with fibrinogen and bind to a different vWF domain (RGD sequence). Unlike the initial vWF-GPIbα interaction, however, the binding of GPIIb/IIIa to vWF has sufficient strength to resist the local shear forces, such that the platelet is firmly arrested on the surface of this tethered ligand (33). If the vWF multimer is of sufficient size, both steps can occur on a single vWF molecule; therefore, the better hemostatic efficiency is of the largest multimers. Accordingly, the initial interaction of GPIbα with vWF has a dual role, one of slowing the platelet and another of inducing the conformational changes that allow it to be cemented to the subendothelial matrix. (15).
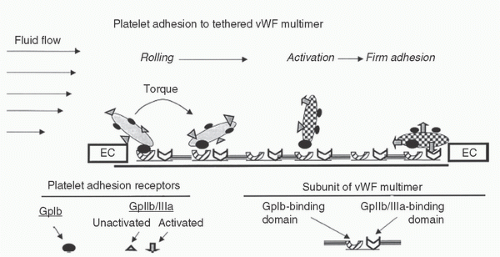 FIGURE 17.2. Platelet adhesion to von Willebrand factor (vWF) at the site of an arterial vessel injury. Schematic representation of the sequence of events in response to platelet adhesion to subendothelium under conditions of high shear stress. The first contact tethers the platelet to an immobilized vWF multimer through the platelet receptor, GPIb. This bond must be formed rapidly, but does not have the strength to hold fast against the high shear present at the vessel wall. The shear rips the platelet free, creating a rolling movement, but the transient binding is sufficient to cause the platelet to become activated. This activation results in a conformational change in a second platelet receptor, GPIIb/IIIa, which allows this receptor to bind to a distinct site on vWF, producing stable, irreversible adhesion to the subendothelium; EC, endothelial cells. (Adapted from Rinder CS. Hematologic effects and coagulopathy. In: Gravlee GP, Davis RF, Stammers AH, et al., eds. Cardiopulmonary bypass-principles and practice. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2008:439–458.) |
Binding of ligands to the GP receptors causes platelets to change their shape, that is, to transform from discoid to spherical, while building lateral finger-like extensions (pseudopods). Simultaneously, platelet fibrinogen receptors (GPIIb/IIIa) are expressed and activated via a conformational change (35), thereby allowing further binding of vWF or fibrinogen as ligands and the formation of a platelet-ligand-platelet matrix.
Moreover, activated platelets release agonists like ADP and serotonin from their granules and thromboxane A2 from their cytosol. Degranulation exerts chemotaxis and stimulation of passing thrombocytes, thereby recruiting them for the amplification of the initial platelet response. The resulting platelet plug built up at the site of endothelial defect is also known as “white thrombus” (33) (Fig. 17.2).
However, the white thrombus generated during primary hemostasis is rather unstable. Crucial for the formation of a stable blood clot is an interaction of activated platelets with the soluble coagulation system (see below). During platelet activation, membrane phospholipids become negatively charged, thereby facilitating the activation of several coagulation factors (predominantly FV and FVIII), binding of the prothrombin complex to the platelet membrane, and finally converting prothrombin to thrombin (33). Thrombin converts fibrinogen to fibrin, which is in turn a potent platelet
activator. Therefore, the activation of further platelets and the simultaneous formation of the fibrin network lead to the formation of the so-called red-thrombus and to strengthening of the blood clot (36).
activator. Therefore, the activation of further platelets and the simultaneous formation of the fibrin network lead to the formation of the so-called red-thrombus and to strengthening of the blood clot (36).
Platelet Defects during CPB
Qualitative and quantitative platelet defects are typically witnessed during CPB, both having the potential to cause significant bleeding (18). During CPB, platelet counts usually decrease by 30% to 50%. Occasionally, this can result in dips below 50,000/µL, which is an accepted transfusion trigger for platelet concentrates in surgical patients (37). However, functional platelet defects elicited by CPB may produce bleeding that requires platelet transfusion despite seemingly adequate platelet counts (15). Additionally, platelet function is frequently restricted by antiplatelet medication.
While hemodilution with acellular priming solutions generally appears to be a plausible explanation for decreases in platelet counts, some further mechanisms deserve consideration in this context. One mechanism most likely to decrease the number of circulating thrombocytes is their adhesion to the circuit surfaces, which are coated with adsorbed fibrinogen, vWF, and fibronectin. Upon activation, platelets express their GPIIb/IIIa receptors (see above), which allows them to adhere to fibrinogen and vWF (19). Further physical factors contributing to CPB-associated thrombopenia include mechanical disruption, shear stress, hypothermia, and sequestration in pulmonary and splenic tissues (38). Moreover, consumptive coagulopathy (19,39) and the formation of platelet conjugates (i.e., platelet-monocyte or platelet-neutrophil) also contribute to decreasing platelet counts (28). While platelet levels drop, a few new platelets enter the circulation either by outpouring from the bone marrow or by recruitment from the spleen. Extracorporeal circulation thus has a profound effect on platelet population, which can become highly heterogenous during CPB (19).
Several alterations in platelet function can occur in the peri-CPB patient, with no single functional defect accounting for the preponderance of bleeding. In general, platelet response to stimulation by agonists gets blunted. Accordingly, higher concentrations of ADP, collagen, thrombin, and other platelet agonists are required to achieve irreversible platelet aggregation. Moreover, the ability to adhere in the presence of high shear is decreased (15).
The emergence of platelet dysfunction appears to be multifactorial with platelet activation being the leading cause. Platelet activation becomes measurable by release of granular contents and essentially results from direct contact with the synthetic material of the CPB circuit. Most likely, this process is mediated by the GPIIb/IIIa receptor (40,41). Within the circuit as well as in the pericardial wound, low concentrations of thrombin—a potent platelet activator—are generated. Most likely, this effect is mediated by activation of protein-C-kinase and the upregulation of P-selectin and interactions with the GPIb receptor (38). In the further course of surgery, platelets also become activated by complement factors (C5b-9), leukotrienes, plasmin, platelet-activating factor (PAF), and different collagenases (19). Hypothermia, heparin dosing, and antagonization of heparin with protamine also contribute to the alterations of platelet counts and function (19). Heparin (which is used as an anticoagulant for CPB) binds to the surface of platelets, thereby causing degranulation of the a-granules (38). Moreover, heparin has been observed to bind to vWF at a site critical for binding to platelet GPIb (42). The neutralization of heparin with protamine leads to a transient decrease of platelet count, which is associated with activation of platelets via the classic pathway of the complement system and the formation of transient aggregates, which are sequestered in the lungs (43).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


