I. INTRODUCTION
A. Heart failure is a complex clinical syndrome characterized by impaired myocardial performance and progressive maladaptive neurohormonal activation of the cardiovascular and renal systems leading to circulatory insufficiency and congestion. Currently, acute heart failure syndromes (AHFS) constitute the most common indication for hospitalization in adults over age 65. With the increasing age of the population, improved survival of patients with acute coronary syndromes, and reduced mortality from other diseases, the incidence and attendant cost of managing patients with heart failure will inevitably increase.
B. Terminology
1. Based on the hemodynamic model,
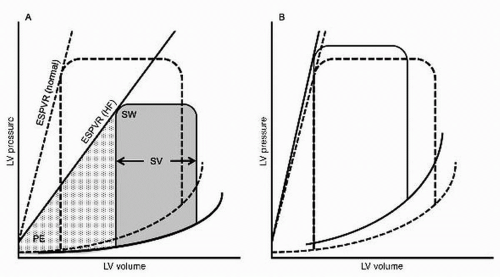
systolic heart failure has been defined by the presence of impaired contractility of the left ventricle, most commonly conveyed in an ejection fraction (EF) of < 40% to 50%. This drop in contractility may be associated with chamber dilation and a decreased stroke volume (
Fig. 8.1). There is a growing appreciation for the limitations of this classification. The threshold for systolic dysfunction is arbitrary and it is now clear that patients with heart failure with preserved EF suffer similar morbidity and mortality. There is substantial variability in EF determinations made by different imaging modalities. Most importantly, EF correlates poorly with symptoms, cardiac indices, and potential response to pharmacotherapy.
2. In practice, heart failure is a bedside diagnosis that is defined by clinical assessment. Patients may have cardiac dysfunction without symptoms, often referred to as
asymptomatic left ventricular (LV) dysfunction. Others may have preserved LV systolic function with typical signs and symptoms of heart failure, best referred to as
heart failure with preserved EF (see
Chapter 9).
3. The major pathophysiologic process in the progression of heart failure is cardiac remodeling, in the form of progressive chamber enlargement with an obligatory reduction in EF. Histopathologically, this is associated with myocyte hypertrophy, apoptosis, and necrosis. Molecular alterations including reexpression of a fetal gene program and alterations in excitation-contraction coupling and regulatory proteins occur.
4. In some cases, myocardial recovery or reverse remodeling is possible with pharmacologic and device therapy.
5. The term congestive heart failure is overused and nonspecific, often being applied to states of hypervolemia unrelated to cardiac dysfunction. Conversely, not all patients with heart failure have signs and symptoms of congestion.
6. The term right heart failure is used to describe patients with predominantly peripheral signs and symptoms of heart failure with a relative paucity of pulmonary congestion.
7. Acute decompensated heart failure or AHFS refer to episodes of acute or subacute deterioration of heart failure due to a wide range of precipitants. The vast majority of these events are marked by systemic and pulmonary congestion.
II. PATHOGENESIS
A. Heart failure is a progressive disorder initiated by some form of myocardial injury. This injury may range from acute disruptions in myocardial function (myocardial infarction or myocarditis) to one of a number of chronic derangements including familial and metabolic cardiomyopathies or chronic volume or pressure loading related to valvulopathies, intracardiac shunts, or systemic hypertension. Regardless of the initial insult, the compensatory mechanisms that may be beneficial acutely ultimately become maladaptive in the chronic phase.
B. Neurohormonal activation
1. Activation of the sympathetic nervous system. Chronic activation of the sympathetic nervous system ultimately results in decreased β-adrenergic receptor responsiveness and decreased norepinephrine stores and sympathetic innervation of the myocardium. Chronically, these changes contribute to myocyte hypertrophy, fibrosis, and necrosis. Extracardiac effects include increased tubular reabsorption of sodium, activation of the renin-angiotensin system (RAS), neurogenic vasoconstriction, and vascular hypertrophy.
2. Activation of the RAS. As heart failure progresses, renal hypoperfusion and sympathetic stimulation of the kidneys result in increased production of renin by the juxtaglomerular apparatus. Renin cleaves circulating angiotensinogen into the biologically inactive angiotensin I, which is subsequently cleaved by angiotensin-converting enzyme (ACE) to the biologically active angiotensin II. Importantly, angiotensin II can be generated in renin and ACE-independent pathways as well. In addition to direct cardiovascular effects, angiotensin II stimulates aldosterone production by the zona glomerulosa of the adrenal cortex, which in turn promotes reabsorption of sodium in exchange for
potassium in the distal nephron. Chronically, aldosterone results in the promotion of hypertrophy and fibrosis in the vasculature and myocardium, endothelial dysfunction, and inhibition of norepinephrine uptake.
3. Other neurohormonal derangements. Inappropriate production of arginine vasopressin has an antidiuretic effect contributing and worsens vasoconstriction. Endothelin, neuropeptide Y, and other peripheral vasoconstrictors further enhance vascular tone.
VI. ETIOLOGY.
It is essential to make every effort to identify the specific etiology of heart failure as it may have implications for management and prognosis. While ischemic cardiomyopathy is by far the most common cause of systolic heart failure, a diverse array of disease states can culminate in this phenotype (
Table 8.2).
A. Ischemic cardiomyopathy is the cause of 60% to 75% of cases of systolic heart failure in industrialized countries. It is defined as cardiomyopathy in the presence of prior extensive myocardial infarction, hibernating myocardium, or severe coronary artery disease. However, the mere presence of obstructive coronary artery disease does not equal ischemic cardiomyopathy as it is possible to have coronary artery disease superimposed with a nonischemic etiology of heart failure. A careful assessment of the coronary anatomy, the burden of ischemia, and the presence of infarcted and viable myocardium must be made and an assessment of the proportionality of these findings to the degree of myocardial dysfunction should be made. The risks and benefits of percutaneous or surgical revascularization should be assessed in all patients with ischemic cardiomyopathy. Extensive observational data have suggested a benefit for coronary artery bypass grafting (CABG) compared with medical therapy alone in moderate to severe LV systolic dysfunction. Registry data suggest that CABG is superior to percutaneous coronary intervention in patients with reduced EF. However, recently released 5-year data from the Surgical Treatment for Ischemic Heart Failure (STICH) trial demonstrated no difference in 5-year mortality for patients with left ventricular ejection fraction (LVEF) < 35% undergoing CABG in addition to optimal medical therapy versus optimal medical therapy alone. Furthermore, a substudy of STICH demonstrated that preoperative viability testing did not effectively predict whether patients derived benefit from CABG. Notably, patients with left main trunk disease and severe angina were excluded from the study and these patients should continue to be treated aggressively with revascularization.
B. Dilated cardiomyopathy. In 20% to 30% of cases of heart failure with systolic dysfunction, the precise etiology is not established and a diagnosis of nonischemic, dilated, or idiopathic cardiomyopathy is made. Patients with dilated cardiomyopathy typically have a better prognosis than their ischemic counterparts.
1. Dilated cardiomyopathy is frequently attributed to the residual effects of subclinical viral myocarditis. Reverse transcription polymerase chain reaction analysis of endomyocardial biopsies from patients with dilated cardiomyopathy demonstrates amplification of viral genomes in approximately two-thirds of cases. Any virus can cause myocarditis, but, owing to its ubiquity, coxsackie B virus is the most epidemiologically important.
2. Familial dilated cardiomyopathy. It is now recognized that 25% to 50% of cases of dilated cardiomyopathy may have a genetic basis. Conditions are typically autosomal dominant and show variable penetrance. A detailed threegeneration family history is essential at the time of initial evaluation. If the family history suggests a genetic predisposition, clinical screening of family members is appropriate and genetic testing can be performed following referral to a genetic counselor. Importantly, only 15% to 25% of presumed familial dilated cardiomyopathies have identifiable genetic alterations.
C. Hypertensive and diabetic cardiomyopathy are seldom considered as stand-alone diagnoses. Progression from LV hypertrophy to overt dysfunction in hypertensive patients (the so-called burnt-out hypertensive heart) most likely results from progressive microvascular ischemia. Hypertension and diabetes also contribute significantly to the development of coronary artery disease and ischemic cardiomyopathy.
D. Cardiotoxic agents. The list of toxins that can produce cardiomyopathy is extensive. Identification of the toxin and removal of the offending agent may halt the progression of or even reverse LV dysfunction.
1. Chemotherapeutic agents. Anthracycline toxicity can cause myocyte destruction and cardiomyopathy. Patients who receive a cumulative doxorubicin equivalent dose of < 400 mg/m
2 are at low risk for this syndrome, while those receiving a cumulative dose > 700 mg/m
2 have an approximately 20% lifetime risk of developing cardiomyopathy. Other cardiotoxic drugs that require careful cardiac monitoring include
cyclophosphamide and
trastuzumab. Trastuzumab
(Herceptin) is now frequently used in the treatment of human epidermal growth factor receptor 2 positive breast cancer and has been associated with a reversible cardiomyopathy in 2% to 7% of patients undergoing treatment. Antiangiogenic drugs such as sunitinib can also cause cardiotoxicity and uncontrolled hypertension.
2. Alcohol consumption is thought to represent a common cause of toxin-mediated cardiomyopathy; however, there is limited observational data on the actual incidence of the cardiomyopathy or the volume of alcohol consumption necessary to induce it. Total abstinence from alcohol may result in complete resolution, whereas continued use is associated with a 3- to 6-year mortality exceeding 50%.
3. Stimulant drugs including cocaine and methamphetamine may result in the development of heart failure via multiple derangements including progressive concentric hypertrophy and recurrent myocardial infarction.
4. Toxin exposures including lead, arsenic, and cobalt can result in progressive myocardial dysfunction.
E. Inflammatory cardiomyopathy (i.e., myocarditis) is discussed in detail in
Chapter 11.
F. Tachyarrhythmia-induced cardiomyopathy can complicate the course of atrial fibrillation, atrial flutter, ectopic atrial tachycardia, and even occult sustained ventricular tachycardia and frequent premature ventricular contractions (> 20% to 30% of beats). In general, it is thought that persistent tachycardia in excess of 110 bpm is required to induce LV dysfunction. This is a critical diagnosis to make, as treatment of the underlying tachyarrhythmia generally results in complete resolution of the cardiomyopathy.
G. Peripartum cardiomyopathy is defined as a dilated cardiomyopathy occurring between the last month of pregnancy and up to 5 months postpartum. The majority of peripartum cardiomyopathy patients improve with standard heart failure pharmacotherapy, with over 50% of patients experiencing complete normalization of cardiac function.
H. Valvular disorders are common causes of heart failure. Aortic regurgitation and mitral regurgitation (MR) result in chronic volume overload and ultimately culminate in dilated cardiomyopathy. Severe aortic stenosis and outflow tract obstruction commonly lead to progressive LV dysfunction (see
Chapters 14 and
15). Surgical correction is the preferred management of severe valvular lesions.
I. Miscellaneous disorders
1. Thyroid disorders
a. Hypothyroidism is common in patients with heart failure. Severe hypothyroidism (i.e., myxedema) may cause decreased cardiac output and heart failure. Bradycardia and pericardial effusion can develop in extreme cases of hypothyroidism.
b. Heart failure may complicate hyperthyroidism, especially in elderly patients with low ventricular reserve. Atrial fibrillation is a common accompanying arrhythmia, occurring in 9% to 22% of patients with thyrotoxicosis. Nonspecific symptoms such as fatigue, weight loss, and insomnia predominate. Previously stable angina may become unstable. Patients treated with amiodarone may develop a wide range of thyroid disorders ranging from abnormal thyroid function tests to overt amiodarone-induced thyrotoxicosis or hypothyroidism. Both conditions can occur in otherwise normal thyroid glands.
2. Thiamine deficiency (beriberi). Although rare in industrialized countries, thiamine deficiency is still prevalent in the developing world. It can also occur in alcoholics or individuals observing fad diets. Wet beriberi includes features of
high-output cardiac failure such as marked edema, peripheral vasodilatation, and pulmonary congestion. The signs and symptoms of dry beriberi include glossitis, hyperkeratosis, and peripheral neuropathy. The laboratory diagnosis is made using decreased RBC transketolase and 24-hour urine thiamine
levels. Severe cases can present with lactic acidosis. Intravenous therapy with 100 mg of thiamine followed by daily oral supplementation can result in dramatic clinical improvement. Chronic use of high-dose diuretics may be complicated by subclinical thiamine deficiency of unknown significance.
3. Other nutritional deficiencies. Carnitine and selenium deficiency may result in dilated cardiomyopathy complicating chronic parenteral nutrition.
4. High-output heart failure from anemia. Acute anemia caused by rapid blood loss is associated with decreased cardiac output due by hypovolemic shock. In contrast, chronic anemia can be associated with symptoms of heart failure due to compensatory mechanisms. These include fluid retention, increased cardiac output, decreased vascular resistance, and increased 2,3-diphosphoglycerate with a resultant rightward shift in the oxyhemoglobin dissociation curve. Moderate degrees of chronic anemia (hemoglobin < 9 g/dL) typically only result in heart failure symptoms in patients with preexisting cardiac disease. Chronic anemia of severe proportions (hemoglobin < 7 g/dL) may result in high-output heart failure even in individuals with normal hearts. Evaluation and management of the underlying cause and supportive care are advised. Thresholds for transfusion depend on the clinical context and rapidity of blood loss. Iron repletion should be considered in iron-deficient patients and in inpatients is most readily achieved with the daily administration of intravenous ferric gluconate 125 mg for 8 to 10 days.
5. While early in its course hemochromatosis may present with restrictive cardiomyopathy, it typically progresses to a mixed or dilated form. Treatment with chelating agents or phlebotomy may improve cardiac function in both primary and secondary forms.
6. Inherited myopathies such as Becker’s and Duchenne’s muscular dystrophies, limb girdle dystrophy, and myotonic dystrophy are associated with dilated cardiomyopathy. Friedreich’s ataxia is most commonly associated with hypertrophic cardiomyopathy, but in rare instances can present with a dilated phenotype. Mitochondrial cardiomyopathies may also present with dilated cardiomyopathy.
7. Cardiac sarcoidosis can present with LV dysfunction with regional hypokinesis or aneurysmal dilatation. It is frequently associated with conduction abnormalities and ventricular tachyarrhythmias. The diagnosis can be supported with stereotypical findings on cardiac MRI and positron emission tomography (PET). The diagnosis is rare in the absence of extracardiac manifestations.
8. Chagas disease caused by the flagellate protozoan Trypanosoma cruzi remains a common cause of heart failure in patients from Latin America. In the chronic symptomatic phase, patients typically present with a syndrome of ventricular dysfunction with regional wall motion abnormalities in the absence of obstructive coronary artery disease. This pattern should prompt T. cruzi titers in patients from endemic regions.
VI. DIAGNOSTIC EVALUATION
A. Laboratory work is used to diagnose potentially reversible causes, identify comorbidities, monitor and correct abnormalities before or during treatment, and assess the disease severity.
1. A
comprehensive metabolic panel should be assessed on initial evaluation and then subsequently based on clinical judgment. Particular attention should be paid to the presence of hyponatremia, which portends a worse prognosis. Hypokalemia is common in the setting of ongoing diuretic therapy. Hyperkalemia can be seen in the context of overaggressive potassium repletion and ongoing treatment with ACE or aldosterone inhibitors or in diabetic patients with
associated type IV renal tubular acidosis. Aside from the pragmatic considerations, many real-world registries have identified elevated blood urea nitrogen (BUN) and creatinine as powerful predictors of outcome. Renal function must also be taken into account when considering therapy with renally excreted drugs. Transaminitis and in some cases a cholestatic pattern of liver function test abnormalities may be seen in the context of right heart failure.
2. Anemia is present in up to 40% of heart failure patients and is associated with increased mortality and functional impairment. While frequently due to anemia of chronic disease, a thorough diagnostic evaluation should be performed.
3. The natriuretic peptides B-type natriuretic peptide (BNP) and N-terminal pro-B-type natriuretic peptide (NT-proBNP) are released in the setting of increased ventricular dilation or wall stress. Normal ranges (BNP < 100 pg/mL; NT-proBNP < 125 pg/mL if age < 75 years and < 450 pg/mL if age ≥75 years) must be interpreted in the context of associated conditions known to alter levels. Increasing age and worsening renal function are associated with increased levels. There is an inverse relationship between natriuretic peptides and body mass index.
a. Screening for heart failure. Although cardiac dysfunction has been associated with elevated natriuretic peptide levels, the sensitivity is relatively low in asymptomatic patients and is highly dependent on the cut-off levels chosen. In general, routine assessment of BNP is not recommended as a screening test for structural heart disease in asymptomatic patients.
b. Diagnosing heart failure. The primary use of natriuretic peptides remains the diagnosis of heart failure in symptomatic patients particularly when the diagnosis is unclear. The high negative predictive value (up to 90%) in this setting allows BNP testing to be useful to rule out a cardiac cause of symptoms. With the growing epidemic of obesity, it is important to remember that normal natriuretic peptide levels may be present in morbidly obese patients with decompensated heart failure.
c. Management of heart failure. While still controversial, there is emerging evidence that serial measurements of natriuretic peptides may be beneficial in guiding outpatient heart failure management and may result in decreased heart failure-related mortality versus usual care.
d. Determining prognosis of heart failure. Data now suggest that natriuretic peptide levels are closely correlated with morbidity and mortality in patients with both established heart failure and other cardiovascular diagnoses (e.g., stable coronary artery disease, acute coronary syndromes, pulmonary hypertension, and atrial fibrillation).
4. Other biomarkers. A growing list of biomarkers assessing systemic inflammation, oxidative stress, extracellular matrix remodeling, and myocyte injury is commercially available or in development. While some of these provide useful prognostic information, it is not clear how to best integrate them into the diagnosis and management of heart failure.
5. Thyroid function testing is warranted for all patients with a new diagnosis of heart failure.
6. Iron studies including ferritin, serum iron, and total iron binding capacity (with calculation of percent transferrin saturation) should be performed to screen for hemochromatosis and occult iron deficiency.