Heart Failure with Normal Ejection Fraction
Carolyn S. P. Lam
Margaret M. Redfield
Heart failure (HF) may be defined as the inability of the heart to pump adequate blood volume relative to the metabolic needs of the body or to do so only at abnormally elevated filling pressures, with resultant congestion. Heart failure is not a single disease entity but, rather, a clinical syndrome associated with a number of cardiac abnormalities which may include impairment of valvular, systolic, diastolic, or pericardial function. The diagnosis of HF is based upon clinical signs and symptoms along with evidence of congestion on the chest radiograph. The syndrome was described and its poor prognostic implications recognized prior to the routine assessment of systolic performance by measurement of the ejection fraction (EF) (1). The most enduring diagnostic criteria established for HF, the Framingham criteria, have never included EF, and yet these criteria have served well over time and consistently identify patients with a high prevalence of cardiovascular disease and a uniformly poor prognosis associated with similar modes of death (2,3).
The predominance of systemic and pulmonary congestion in the clinical presentation of HF first directed investigators and clinicians to consider HF as a disorder of volume regulation, the so-called cardiorenal paradigm of HF (4). As contrast ventriculography, radionuclide ventriculography, and echocardiography became available, the syndrome of HF began to be equated with a reduced EF and treatment strategies focused on HF as a syndrome of contractile dysfunction. This has been referred to as the hemodynamic paradigm of HF. During this era, certain rare conditions were recognized to produce the HF syndrome despite preserved EF. However, the possibility that large numbers of patients with HF may have normal EF (HFnIEF) was never entertained.
Because numerous studies have now demonstrated that HFnIEF is very common, the reasons why it was not previously recognized are unclear. Referral bias may play a major role as methods to assess EF were first available to academic and referral centers. Such centers serve a younger and more predominantly male population, a subgroup with a lower prevalence of HFnIEF. Alternatively, it is also very possible that the prevalence of HFnIEF has increased over time, leading to its widespread recognition only as its prevalence increased. Unfortunately, the impact of referral bias and secular trends on the type of HF patients are developing (reduced versus normal EF) has never been studied, but several factors could contribute to an increased prevalence.
The percent of the population over 65 years of age has increased both due to the baby boom phenomenon and to a reduction in mortality from noncardiovascular and cardiovascular causes. Because HFnIEF (even more so than HF in general) is a disease of the elderly, the increased number of older persons and, particularly, older persons with cardiovascular disease could contribute to increases in prevalence of HFnIEF. Access to cardiologists for older persons has also increased and could contribute to increases in recognition of HFnIEF in recent years. Finally, one could also speculate that advances in the treatment of hypertension and coronary disease have modified but not prevented vascular and cardiac remodeling and dysfunction in such a manner as to preserve EF but not prevent vascular stiffening, diastolic dysfunction, or volume overload—all mechanisms postulated to lead to HFnIEF.
Whatever the reason, the fact that academic cardiologists and HF specialists had not recognized or been exposed to large numbers of patients with HFnIEF led to
considerable skepticism regarding HFnIEF, despite growing epidemiological evidence of its importance. Controversy regarding the existence and prevalence of HFnIEF has largely but not completely abated. Many questions persist due to our lack of knowledge regarding the pathogenesis, natural history, and appropriate therapy for HFnIEF. For example, how HFnIEF fits into the neurohumoral paradigm of HF remains to be defined. There are few data concerning the neurohumoral profile of HFnIEF or the impact of neurohumoral antagonists on outcomes in HFnIEF. Happily, these issues are beginning to be addressed. This chapter summarizes what is currently known regarding HFnIEF, but the authors are careful to emphasize that our understanding of this syndrome is incomplete and will continue to evolve as the field advances.
considerable skepticism regarding HFnIEF, despite growing epidemiological evidence of its importance. Controversy regarding the existence and prevalence of HFnIEF has largely but not completely abated. Many questions persist due to our lack of knowledge regarding the pathogenesis, natural history, and appropriate therapy for HFnIEF. For example, how HFnIEF fits into the neurohumoral paradigm of HF remains to be defined. There are few data concerning the neurohumoral profile of HFnIEF or the impact of neurohumoral antagonists on outcomes in HFnIEF. Happily, these issues are beginning to be addressed. This chapter summarizes what is currently known regarding HFnIEF, but the authors are careful to emphasize that our understanding of this syndrome is incomplete and will continue to evolve as the field advances.
Nomenclature
There is controversy over proper nomenclature to use for HFnIEF. Alternative terms that have been used to divide patients with HF into two groups have included diastolic versus systolic HF, HF with preserved versus reduced systolic function, and HF with normal versus reduced EF. These three sets of terms were used interchangeably in the literature to date, but controversy over the most appropriate terminology arose as interest in the field expanded.
Diastolic Heart Failure
The term diastolic heart failure (DHF) was proposed to underscore the fundamental difference in pathophysiological mechanisms believed to cause HF in these patients, as opposed to patients with HF and reduced EF (HF↓EF), which was referred to as systolic heart failure (SHF). The term implies that HFnIEF is due to diastolic dysfunction. However, in practice there is frequently no assessment of diastolic function in patients with HFnIEF. Diastolic function is difficult to measure and there are no clear diastolic dysfunction criteria considered pathognomonic for HFnIEF. Furthermore, patients with HFnIEF often have coexistent abnormalities in systolic function, and diastolic dysfunction clearly contributes to the pathophysiology of HF↓EF. With this overlap, the term is less useful to clearly separate HF patients into two groups. The SHF/DHF distinction is easy to use and is well-recognized and supported by some studies. Although some investigators argue for a major role of diastolic dysfunction in most, if not all, patients with HFnIEF (5), others argue that the term DHF should not be applied unless or until diastolic dysfunction is proven to be the predominant mechanism in all patients with HFnIEF (6).
Heart Failure with Preserved Systolic Function
This term is also suboptimal because it is still unclear as to whether or not systolic function is indeed preserved in patients with HFnIEF. Various reports have suggested increased, unchanged, or reduced systolic function in patients with HFnIEF depending on which index is used to assess systolic function. In addition, a normal EF does not necessarily mean that myocardial contractility is normal.
Heart Failure with Normal Ejection Fraction
EF measurement makes no assumptions regarding pathophysiological mechanisms; it is clinically useful and has been the basis of inclusion for most HF clinical studies. This term has been used in the revised American College of Cardiology/American Heart Association (ACC/AHA) HF guidelines (7). The term HFnIEF will therefore be used for the purposes of this chapter. However, it should be noted that EF is a continuous variable with a normal distribution within the population, and the threshold value to define normal versus reduced EF is arbitrary. There is no clear consensus in published literature regarding an optimal cut-off value to discriminate between patients in whom systolic dysfunction is the primary mechanism responsible for the patient’s HF symptoms. Indeed, as diastolic dysfunction contributes potently to symptoms and prognosis among patients with reduced EF, this terminology does not clearly distinguish the pathophysiological mechanisms involved. In support of this terminology, patients with an EF <40% and HF symptoms have been proven to derive symptomatic and survival benefit from a number of therapies, whereas benefit is unclear in those with EF >40%. While consensus seems to be building toward use of an EF ≥50% to designate HFnIEF, the proper designation of and treatment approach to patients with borderline (40% to 50%) EF adds to the complexity of the classification.
Ideally, the HF population should be divided according to pathophysiology, which, in turn, will form the basis of differential therapeutic strategies. Future nomenclature may become complicated as multiple pathophysiological mechanisms may become defined in HFnIEF. A greater understanding of the syndrome should ultimately lead to selection of terminology which reflects the underlying pathophysiological mechanisms and aids in investigating and developing therapeutic modalities.
Do Patients with Hfnief have Heart Failure?
Clinical Features
Early studies compared clinical features between HF patients with normal versus reduced EF. Both Dougherty et al. (8) and Echeverria et al. (9) concluded from their respective series of HF patients that neither history, physical examination, nor chest roentgenographic findings were able to distinguish patients with normal versus reduced EF. Similarly, Aguirre et al. (10) reported that no single clinical parameter (e.g., exertional dyspnea, orthopnea, paroxysmal nocturnal dyspnea, edema, rales, jugular venous distension, S3 or S4 gallop) was found to discriminate between HFnIEF or HF↓EF in their series of patients with HF. Of the established clinical diagnostic criteria for HF (Framingham, Boston, or Duke criteria), none had a high sensitivity or specificity for discerning which HF patients had reduced
EF (11). More recent publications also corroborate these findings (12) and establish that decompensated HF with normal versus reduced EF are clinically indistinguishable.
EF (11). More recent publications also corroborate these findings (12) and establish that decompensated HF with normal versus reduced EF are clinically indistinguishable.
Pathophysiological Features
The pathophysiological domains that characterize classic HF were studied by Kitzman et al. (13) in stable outpatients with a clinical diagnosis of HF and normal or reduced EF and age- and sex-matched healthy controls. The four key domains examined included left ventricular (LV) structure and function, exercise performance, neuroendocrine function, and quality of life (Table 7-1 and Fig. 7-1). Patients with HFnIEF were shown to have similar pathophysiological characteristics compared to patients with HF↓EF, including severely reduced exercise capacity, neuroendocrine activation, and impaired quality of life. These similar characteristics were found despite a unique LV structural and functional profile in the patients with HFnIEF, including normal LVEF, normal LV volume, and an increased LV mass to volume ratio. It must be noted that this study was performed in stable outpatients; quality of life, exercise parameters, and neurohumoral function may be more severely perturbed in more symptomatic patients studied in a hospital setting. Nonetheless, by demonstrating perturbations in these key components of the HF syndrome, the validity of the term HF in patients with HFnIEF is therefore established. This is one of the first studies to demonstrate activation of the sympathetic nervous system and the naturietic peptide system in HFnIEF. Unfortunately, characterizations of the renin-angiotensin-aldosterone system, endothelin, and cytokines in HFnIEF were not provided and remain important areas for future studies.
Epidemiologic Features
As reviewed in more detail later, several epidemiological and clinical studies have compared mortality rates in patients with HFnIEF and HF↓EF (14,15). On aggregate, the studies suggest that survival rates are nearly similar between these groups of HF patients; mortality differences, when present, are relatively minor. It should also be emphasized that compared to age-matched controls, mortality risk is clearly higher in patients with HFnIEF. As for morbidity, available comparative studies of hospital readmission rates in patients with HFnIEF versus reduced EF indicate that the readmission rates are almost identical. In a prospective study of 413 patients hospitalized for HF, Smith et al. (16) compared hospital readmission and changes in functional status in HF patients with preserved (EF ≥40%) versus depressed EF. After 6 months, the rates of functional decline were similar among those with preserved and depressed EF (30% versus 23%, respectively; p = 0.14). After adjusting for demographic and clinical covariates, there was no difference in the risk of readmission (HR 1.01; 95% CI 0.72 to 1.43; p = 0.96) or the odds of functional decline or death (OR 1.01; 95% CI 0.59 to 1.72; p = 0.97) between HF patients with preserved versus reduced EF. Thus, HFnIEF confers a considerable burden on patients with the risk of readmission, disability, and symptoms subsequent to hospital discharge, comparable to that of HF patients with reduced EF. These studies establish that patients with HFnIEF do indeed have HF, with similar implications for their quality and quantity of life as seen with HF↓EF.
Finally, it should be recognized that the diagnostic criteria for HF are somewhat nonspecific and other conditions can mimic HF clinically, particularly in elderly patients. Thus, it is likely that misdiagnosis as HF occurs in a subset of patients labeled as HFnIEF. However, the studies summarized throughout this chapter suggest that this is not common.
Pathophysiology of HFnlEF
As established in patients with systolic dysfunction, HF is a progressive syndrome characterized by complex cardiac and
systemic adaptations that vary over time. In patients with HF↓EF, a regional or global myocardial insult leads to systolic and diastolic dysfunction, LV remodeling (hypertrophy, fibrosis, and dilatation), local and systemic neurohumoral and cytokine activation, impairment of renal hemodynamic and excretory function, and vascular dysfunction. Prognostic hemodynamic subgroups are based on relative derangement in cardiac output, filling pressures, and systemic vascular resistance. Chronic elevation of left atrial pressures leads to right heart failure. Progression is modulated by comorbidities, particularly coronary and renal disease. These insights have directed development of therapeutic strategies ultimately proven in clinical trials. This process is incomplete, and genomic and proteomic paradigms for HF pathophysiology will continue to evolve.
systemic adaptations that vary over time. In patients with HF↓EF, a regional or global myocardial insult leads to systolic and diastolic dysfunction, LV remodeling (hypertrophy, fibrosis, and dilatation), local and systemic neurohumoral and cytokine activation, impairment of renal hemodynamic and excretory function, and vascular dysfunction. Prognostic hemodynamic subgroups are based on relative derangement in cardiac output, filling pressures, and systemic vascular resistance. Chronic elevation of left atrial pressures leads to right heart failure. Progression is modulated by comorbidities, particularly coronary and renal disease. These insights have directed development of therapeutic strategies ultimately proven in clinical trials. This process is incomplete, and genomic and proteomic paradigms for HF pathophysiology will continue to evolve.
Table 7-1 Key Pathophysiological Domains in Heart Failure with Normal or Reduced Ejection Fraction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
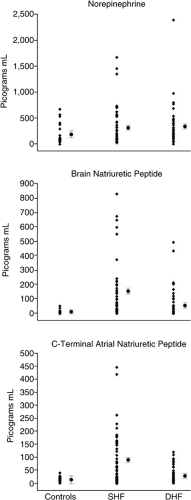 Figure 7-1 Neuroendocrine function in healthy subjects without heart failure (Controls) and in stable outpatients with HF↓EF (SHF) or HFnIEF (DHF). (From Kitzman DW, Little WC, Brubaker PH, et al. Pathophysiological characterization of isolated diastolic heart failure in comparison to systolic heart failure. JAMA. 2002;288(17):2144–2150 .) |
Whether patients with HFnIEF share most pathophysiological mechanisms with HF↓EF remains to be established. The first step in this process is to understand the fundamental abnormality in ventricular function that causes (or at least accompanies) the clinical syndrome. Three theories regarding the perturbations in cardiovascular function responsible for HFnIEF have emerged. Importantly, each theory is not exclusive of the others. Multiple mechanisms may contribute in any given patient although it may be that one is predominant in some patients and others predominate in other patients. These theories are: (a) HFnIEF is due to intrinsic diastolic dysfunction (impaired relaxation and increased diastolic stiffness); (b) HFnIEF is due to increased vascular and ventricular systolic stiffness associated with enhanced sensitivity to volume and load/stress-induced diastolic dysfunction; and (c) HFnIEF is due to ventricular remodeling characterized by LV dilatation with volume-dependent elevation of filling pressures unassociated with significantly altered diastolic stiffness or impaired relaxation. Each of these theories will be discussed in turn.
HFnIEF Is Due to Intrinsic Diastolic Dysfunction
Consideration of the role of diastolic dysfunction in HFnIEF mandates clear understanding of diastolic function. Normal diastolic function allows the ventricle to fill adequately during both rest and exercise without an abnormal increase in diastolic pressures. The phases of diastole are isovolumic relaxation and the filling phase. The filling phase is divided into early rapid filling, diastasis, and atrial systole. Early rapid filling contributes 70% to 80% of LV filling in normal individuals, and this contribution diminishes with age and a variety of disease states. Early diastolic filling is driven by the left atrial (LA) to LV pressure gradient, which is dependent on a complex interplay of factors: myocardial relaxation, passive elastic properties of LV, LV elastic recoil, LV contrctile state, LA pressures, ventricular interaction, pericardial constraint, elasticity of LA, pulmonary veins, and mitral orifice area. Diastasis is the period of diastole when the LA and LV pressures are usually nearly equal. It contributes less than 5% of the LV filling and its duration shortens with tachycardia. In normal subjects atrial systole contributes 15% to 25% of LV diastolic filling without raising the mean left atrial pressure. This contribution depends on the PR interval, atrial inotropic state, atrial preload, atrial afterload, autonomic tone, and heart rate (17).
Assessment of diastolic function is obtained with invasive diagnostic techniques (cardiac catheterization) or noninvasive imaging modalities (echocardiography radionuclide angiography). Recent reviews have provided
extensive analyses of the invasive and noninvasive assessment of LV diastolic function (17,18,19,20,21). Features of the invasive and Doppler echocardiographic assessment of diastolic function will be further discussed.
extensive analyses of the invasive and noninvasive assessment of LV diastolic function (17,18,19,20,21). Features of the invasive and Doppler echocardiographic assessment of diastolic function will be further discussed.
Left Ventricular Relaxation
While diastolic function is influenced by all the factors referred to above, its primary determinants are LV relaxation and LV compliance. LV relaxation is an active, energy-dependent process that begins during the ejection phase of systole and continues through isovolumic relaxation and the rapid filling phase. It is considered that the LV relaxation is under the triple control of systolic load, myofiber inactivation, and the uniformity of the distribution of load and inactivation in space and time (22,23,24). The time constant of relaxation, known as tau, describes the rate of decrease of LV pressure decay during isovolumic relaxation. It requires cardiac catheterization with high-fidelity manometer-tipped catheters for precise determination. The pressure data during the time from end-systole (peak – dP/dt) to onset of LV filling (determined from LA to LV crossover pressure or estimated as 5 mm Hg above the end-diastolic pressure) are fit to a variety of equations to calculate the time constant of isovolumic relaxation. The equations are used make different assumptions regarding the LV minimum pressure obtained. The Weiss equation assumes a zero asymptote or that the LV minimum pressure must be positive. The so-called logistic equation lets the data drive the asymptote and allows for negative LV minimum pressures as has been documented in animal studies. Another equation fits the data to a linear rather than exponential relationship by calculating the slope of the relationship between the derivative of LV pressure and LV pressure over the range of isovolumic relaxation. While the data do not fit this equation as well as the exponential relationships, this method has the advantage of being more independent of preload because the slope of the relationship does not change depending on the LA pressure. While much emphasis has been placed on the method used to derive tau, all methods have been used in clinical studies. With all methods, the larger the value for tau, the more impaired the relaxation.
While LV relaxation may become impaired in the presence of a number of cardiovascular diseases, the role of impaired relaxation in contributing to increased LA pressures at rest is unclear. However, recent data in elderly hypertensive canines (25) and in humans (5) provide evidence that impaired relaxation delays the time to LV minimum pressure well into mid- and even late-diastole and contributes to the mean LA pressures, particularly during the hypertensive episodes. With exercise, there is an increase in venous return and a decrease in the diastolic filling period due to the increase in heart rate. During exercise, the normal ventricle relaxes faster and to a lower minimal pressure, allowing an increase in diastolic filling without an increase in LA pressure, as elegantly demonstrated in conscious, chronically instrumented canines (Fig. 7-2) (26,27,28). This process is mediated by enhanced sympathetic tone. In HF↓EF, the acceleration of LV relaxation with exercise is absent and, therefore, the increased early diastolic filling during exercise occurs at the expense of increased LA pressures. Thus, in patients with impaired LV relaxation, exertional symptoms may occur due to an inability to augment relaxation with exercise. More recently, similar findings have been reported in patients with HFnIEF where blood pressure, tau, and filling pressures increased dramatically with exercise (Fig. 7-3) (29). LV elastic recoil results from release of the potential energy present in the shortened myocardial fibers at the end of systole and is closely related to the level of systolic function. This process has been demonstrated in both filling and nonfilling ventricles. Potentially, elastic recoil could augment the speed of LV pressure fall such that pressure decay may proceed normally even in the presence of abnormalities of myofiber inactivation. Such a situation has been postulated to occur in hypertrophic cardiomyopathy but it remains unclear as to whether elastic recoil preserves the speed of LV chamber relaxation despite myocardial diastolic dysfunction in the more common causes of HFnIEF (17).
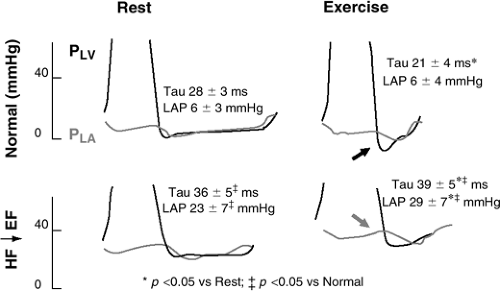 Figure 7-2 Left ventricular (PLV) and left atrial (PLA) pressure tracings in conscious, chronically instrumented canines before (Normal) and after (HF↓EF) induction of severe systolic dysfunction by chronic rapid right ventricular pacing. In Normals, with exercise, relaxation is enhanced with faster (decreased tau) and more complete (lower LV minimum pressure, black arrow) relaxation. This increases the LA to LV pressure gradient in early diastole, allowing enhanced filling without increases in PLA. In contrast, after production of HF↓EF, relaxation does not improve with exercise and the LA to LV pressure gradient (and thus LV filling) can only be increased at the expense of increased PLA. (Modified from Cheng CP, Noda T, Nozawa T, et al. Effect of heart failure on the mechanism of exercise-induced augmentation of mitral valve flow. Circ Res. 1993;72:795–806 .) |
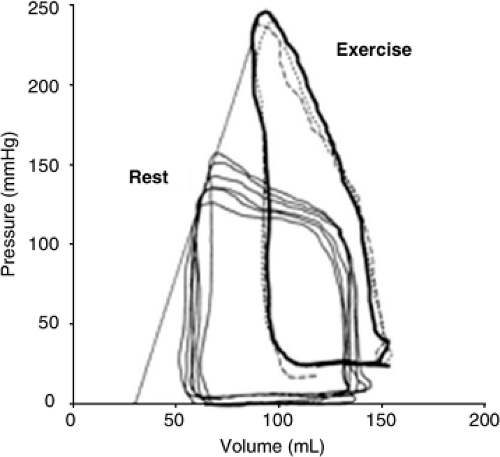 Figure 7-3 Invasive pressure volume data from a patient with HFnIEF before and during hand grip exercise, revealing a hypertensive response concordant with the high basal end-systolic elastance (Ees) and elevated diastolic pressure during stress. On average, end-systolic pressure rose to 200.5 ± 12.3 mm Hg, EDP to 32.3 ± 8.6 mm Hg, and relaxation time to 85.7 ± 23.1 milliseconds (all p <0.05 versus baseline). (Modified from Kawaguchi M, Hay I, Fetics B, Kass DA. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction: implications for systolic and diastolic reserve limitations. Circulation. 2003;107(5):714–720 .) |
Left Ventricular Diastolic Stiffness
Compliance is defined as the relationship between the change in stress and the resulting strain. The inverse of compliance is stiffness or elastance. The elastance of the LV varies over the cardiac cycle (time-varying elastance), and both the end-systolic and end-diastolic elastance are defined by the relationship between pressure and volume as volume is altered (Fig. 7-4) (19). Thus, stiffness is defined by the pressure-volume relationship measured at variable preloads at end-systole (reflective of systolic performance, Ees) and end-diastole (reflective of diastolic stiffness or elastance, Ed). These relationships are altered in the presence of systolic or diastolic function. In terms of LV diastolic function, the diastolic LV stiffness is related to the rate of LV pressure change (measured at end-diastole), which occurs with a given change in LV volume. This relationship is curvilinear in the normal LV. The curvilinear nature of the normal end-diastolic pressure-volume relationship means that the normal LV becomes stiffer and more resistant to filling as the LV diastolic volume is increased.
The effective or operant compliance of the LV is defined as the change in volume and pressure during filling with a single cardiac cycle at any particular end-diastolic volume (Fig. 7-5). A ventricle will have a different operant compliance at different places on its end-diastolic pressure-volume relationship. The operant compliance is influenced not only by the intrinsic myocardial stiffness but also by continued relaxation and recoil, pericardial constraint, ventricular interaction, intrathoracic pressures, and coronary turgor. The intrinsic myocardial stiffness is related to the structure of the LV wall and the viscoelastic forces of the functioning myocardium. Evidence of decreased LV compliance has been demonstrated in patients with hypertrophic cardiomyopathy (30), hypertensive LV hypertrophy (31); acute ischemia (32), valvular disease (33), and restrictive cardiomyopathy (34) and can be inferred in any patient when filling pressures are increased in the presence of normal ventricular volume. While the slope of the curvilinear end-diastolic pressure-volume relationship increases as the compliance decreases, it is also possible to observe an upward shift of the relationship without a change in the slope of the relationship. This has been referred to as decreased distensibility by some investigators and has the same effect as altered compliance with increased pressures needed to maintain the same LV filling volume. However, the mechanisms (extrinsic forces versus intrinsic LV diastolic dysfunction) that mediate such upward shifts are less clear.
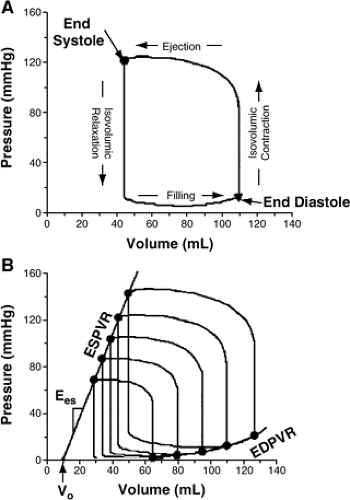 Figure 7-4 A. The four phases of the cardiac cycle are readily displayed on the pressure-volume loop, which is constructed by plotting instantaneous pressure versus volume. This loop repeats with each cardiac cycle and shows how the heart transitions from its end-diastolic state to the end-systolic state and back. B. With a constant contractile state and afterload resistance, a progressive reduction in ventricular filling pressure causes the loops to shift toward lower volumes at both end-systole and end-diastole. When the resulting end-systolic pressure-volume points are connected, a reasonably linear end-systolic pressure-volume relationship (ESPVR) is obtained. The linear ESPVR is characterized by a slope (Ees) and a volume axis intercept (Vo). In contrast, the diastolic pressure-volume points define a nonlinear end-diastolic pressure-volume relationship (EDPVR). (From Burkhoff D, Mirsky I, Suga H. Assessment of systolic and diastolic ventricular properties via pressure-volume analysis: a guide for clinical, translational, and basic researchers. Am J Physiol. (Heart Circ Physiol.) 2005;289(2):H501–H512 .) |
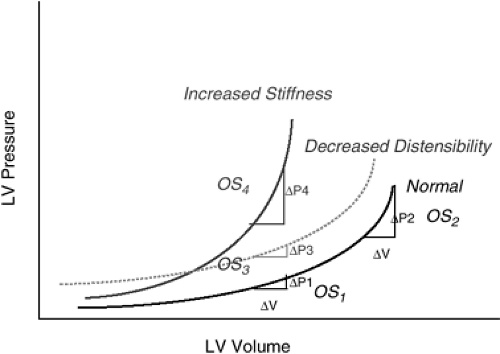 Figure 7-5 End-diastolic pressure-volume relationships showing normal diastolic stiffness, increased stiffness, and decreased distensibility. The operant stiffness (OS) can be altered by moving upward and rightward (OS1 to OS2) on the normal end diastolic pressure-volume relationship (EDPVR), which occurs in the absence of altered diastolic function. In contrast, if the intrinsic diastolic properties of the ventricle are altered (increased stiffness), the operant stiffness will also be increased (OS1 to OS4). In the presence of decreased distensibility, where the end-diastolic pressures are higher at any volume but the slope of the EDPVR is unchanged, the end-diastolic pressures will be higher than in Normals at any given volume, but the operant stiffness will not be different than in Normals. |
Definition of Diastolic Dysfunction
While no standard definition for diastolic dysfunction exists, it may be described as a condition in which higher than normal LV filling pressures are needed to maintain a normal LV volume and cardiac output. It has been postulated that the natural history of diastolic dysfunction progresses through a spectrum characterized by progressive impairment of LV relaxation, which then becomes associated with progressive reduction in LV compliance and elevation in filling pressures (Fig. 7-6) (20,35). This evolution has been documented in experimental dilated cardiomyopathy using invasive assessment of LV relaxation and compliance (36) as well as in humans with the rapidly progressive infiltrative cardiomyopathy associated with amyloid deposition using noninvasive assessment of diastolic function (34,37). The natural history of diastolic dysfunction is likely highly variable in regards to the rate of progression and heavily influenced by the nature of the underlying disease state. The progression of diastolic abnormalities associated with progression to HFnIEF in patients with hypertensive heart disease or coronary disease has yet to be established in longitudinal studies with sequential assessment of diastolic function over time, and this proposed natural history remains highly plausible but as yet not definitively proven to be operative during the natural history of most or all patients who develop HFnIEF.
Evidence for Diastolic Dysfunction in HFnIEF
Several studies support the concept that intrinsic diastolic dysfunction is present and mediates elevated filling pressures in HFnIEF. Studies implicating a role for impaired relaxation and increased diastolic stiffness will be considered next.
Impaired relaxation is present in patients with HFnIEF and contributes to elevated filling pressure at rest or with exercise. When considering whether relaxation is impaired in HFnIEF, comparison to age-matched controls is important because relaxation becomes more impaired with age. If impaired relaxation contributes significantly to the pathophysiology of HFnIEF, some investigators would maintain that relaxation must be perturbed beyond what is seen in asymptomatic age-matched patients. Zile et al. characterized tau in patients with HFnIEF and reported it to be elevated (59 ± 14 milliseconds, n = 47) as compared to age-matched controls (35 ± 14 milliseconds, n = 10, p = 0.01) (5,38). This study also demonstrated that impaired relaxation contributed to the elevation of early (and thus, mean) diastolic pressures.
In contrast, a much smaller study did not find significant differences in tau between HFnIEF patients (n = 10) and age-matched controls (n = 9) at rest (29) but did show dramatic increases in systolic blood pressure, diastolic pressures, and tau with exercise in HFnIEF patients but not age-matched controls (Fig. 7-3). These authors speculated that while significant differences in baseline relaxation were not present in HFnIEF patients, these patients had advanced central aortic and LV systolic stiffening, which magnified stress-induced load and load-dependent diastolic dysfunction, resulting in elevated filling pressures with exercise. This study raises an important point, suggesting that in some HFnIEF patients, resting diastolic function may not be dramatically altered, but exercise or other stressors (hypertensive episodes, volume overload) can produce exaggerated worsening of diastolic function, resulting in stress-induced elevation of filling pressures not seen in asymptomatic age-matched controls.
The concept of differential responses to exercise in HFnIEF is supported by several earlier studies that examined the response to exercise in different ways. Sobue et al. looked at the effect of exercise on filling pressures in patients with normal EF referred to angiography for evaluation of chest pain (39). While these were not described specifically as HFnIEF patients, the findings are pertinent. In patients without coronary disease or LV hypertrophy, pulmonary capillary wedge pressure was normal at rest and at peak exercise (Table 7-2). In patients without coronary disease but with LV hypertrophy, pulmonary capillary wedge pressure was normal at rest but was elevated at peak exercise. In patients with coronary disease, pulmonary capillary wedge pressure was normal at rest but elevated at peak exercise, more so in patients with coronary disease and LV hypertrophy. These data do not define the relative contribution of impaired relaxation or compliance to the elevation of filling pressures but do demonstrate that ventricular hypertrophy and coronary disease (both common in HFnIEF) may not perturb resting filling pressures but can produce exercise-induced diastolic dysfunction and symptoms.
Cuocolo et al. demonstrated that patients with hypertension who had ventricular hypertrophy and impaired relaxation (based on Doppler evaluation) had an inability to enhance filling with exercise, resulting in a flat ejection
fraction response to exercise and suggesting that an inability to enhance relaxation with exercise may contribute to exercise-induced symptoms (40). An elegant study by Kitzman et al. compared exercise pressures and volumes in previously treated outpatients with HFnIEF (most of whom had hypertensive heart disease) and age-matched controls. In this population, none had coronary disease (41). The HFnIEF patients had (on average) normal pulmonary capillary wedge pressures and LV volumes at rest, but with exercise developed dramatic increases in pulmonary capillary wedge pressure unassociated with increases in end-diastolic volume (Fig. 7-7). In these patients, the systolic blood pressures with exercise were not different from controls, suggesting that the exercise-induced diastolic dysfunction was not strictly load-dependent. This study suggests that the Frank-Starling mechanism is impaired in patients with HFnIEF and is consistent with the study of Cuocolo et al. However, this study did not define the relative importance of impaired relaxation versus decreased compliance in mediating the abnormal response to exercise.
fraction response to exercise and suggesting that an inability to enhance relaxation with exercise may contribute to exercise-induced symptoms (40). An elegant study by Kitzman et al. compared exercise pressures and volumes in previously treated outpatients with HFnIEF (most of whom had hypertensive heart disease) and age-matched controls. In this population, none had coronary disease (41). The HFnIEF patients had (on average) normal pulmonary capillary wedge pressures and LV volumes at rest, but with exercise developed dramatic increases in pulmonary capillary wedge pressure unassociated with increases in end-diastolic volume (Fig. 7-7). In these patients, the systolic blood pressures with exercise were not different from controls, suggesting that the exercise-induced diastolic dysfunction was not strictly load-dependent. This study suggests that the Frank-Starling mechanism is impaired in patients with HFnIEF and is consistent with the study of Cuocolo et al. However, this study did not define the relative importance of impaired relaxation versus decreased compliance in mediating the abnormal response to exercise.
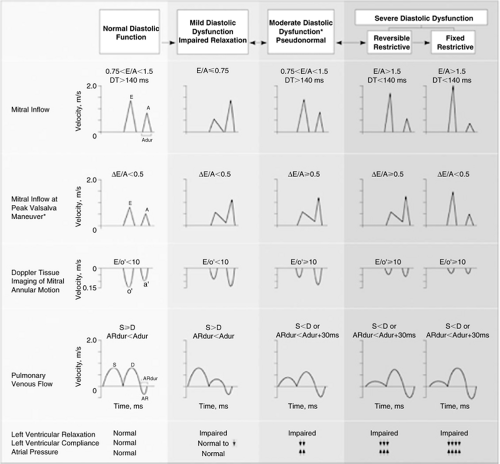 Figure 7-6 The proposed progression of diastolic function abnormalities with correlation of invasively measured diastolic properties (text at bottom) and the corresponding Doppler echocardiographic parameters. (From Redfield MM, Jacobsen SJ, Burnett JC, Jr., et al. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289(2):194–202 .) |
Doppler echocardiography also provides supporting evidence for a role of impaired relaxation to contribute to stress-induced symptoms in HFnIEF. In patients with impaired relaxation, early filling is decreased and the contributions of diastasis flow and atrial contraction are magnified (Fig. 7-8, right panel). In these patients, the entire diastolic filling
period is needed to fill the ventricle, even at slow heart rates. During exercise, the increased venous return and shortened diastasis period will mandate the need for elevation of filling pressures to maintain filling with exercise, contributing to the elevation of filling pressures and inability to augment filling demonstrated in the studies which measured pressures and/or volumes with exercise (40,41) in HFnlEF.
period is needed to fill the ventricle, even at slow heart rates. During exercise, the increased venous return and shortened diastasis period will mandate the need for elevation of filling pressures to maintain filling with exercise, contributing to the elevation of filling pressures and inability to augment filling demonstrated in the studies which measured pressures and/or volumes with exercise (40,41) in HFnlEF.
Table 7-2 Effect of Coronary Artery Disease and Left Ventricular Hypertrophy on the Effect of Exercise on Filling Pressures | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
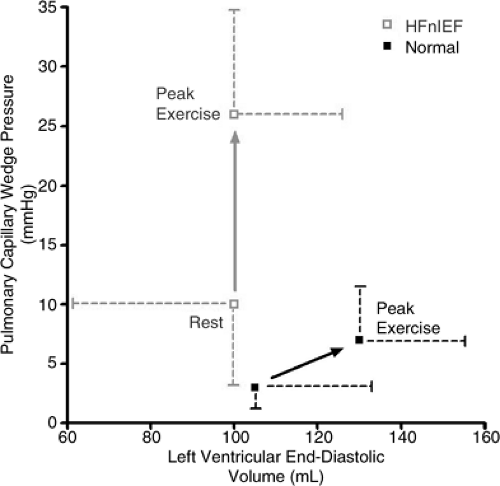 Figure 7-7 Rest and exercise pulmonary capillary wedge pressure (PCWP) and LV volume in normal subjects and patients with HFnIEF. Resting PCWP was higher in HFnIEF but the average was within the normal range. With exercise, the HFnIEF patients could not augment LV volume despite marked elevation in PCWP. In contrast, normal subjects markedly augmented LV volume during exercise without abnormal increases in PCWP. (From Kitzman DW, Higginbotham MB, Cobb FR, et al. Exercise intolerance in patients with heart failure and preserved left ventricular systolic function: failure of the Frank-Starling mechanism. J Am Coll Cardiol. 1991;17(5):1065–1072 .) |
Contribution of impaired relaxation to another common mode of decompensation for HFnlEF patients is also evidenced by the enhanced dependence on atrial contraction for filling. In one of the few studies that carefully examined factors contributing to decompensation in patients with HFnlEF, nearly 30% of hospitalizations for acutely decompensated HFnlEF were due to new onset rapid atrial fibrillation (42). Exacerbation of symptoms with atrial arrhythmias is recognized and included in proposed diagnostic criteria for HFnlEF (43). The acute reduction in filling associated with loss of atrial systolic activity in atrial fibrillation, coupled with the resistance to early diastolic filling due to impaired relaxation, will again mandate acute elevation of filling pressures (44,45) and contribute to acute episodes of HFnlEF.
In contrast, in normal individuals with normal relaxation and brisk, early diastolic filling, dependence on diastasis flow and atrial filling is minimal (Fig. 7-8, left panel) and rapid atrial fibrillation is much less likely to induce acute pulmonary edema. This scenario is also supported by modeling studies which suggest that elevation of tau to levels observed in HFnlEF (60 milliseconds), while not affecting resting filling pressures, will produce dramatic increases in filling pressures with tachycardia
(46). In this experiment, compliance was held constant, suggesting that relaxation impairment alone can contribute to elevation of mean left atrial and pulmonary venous pressures with tachycardia.
(46). In this experiment, compliance was held constant, suggesting that relaxation impairment alone can contribute to elevation of mean left atrial and pulmonary venous pressures with tachycardia.
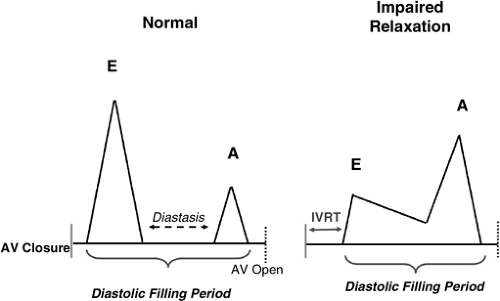 Figure 7-8 Insights into the importance of impaired relaxation in influencing atrial pressures from Doppler echocardiography. Transmitral inflow velocity profiles in normal diastolic function and in the presence of impaired relaxation are shown. In normal subjects (left), the diastolic filling period is longer due to a shorter isovolumic relaxation time (IVRT). Most filling occurs in early diastole due to brisk LV relaxation, which enhances the atrial-to-LV pressure gradient in early diastole. Thus, there is little filling during diastasis and minimal contribution from atrial contraction. In the presence of markedly impaired relaxation (right), the diastolic filling period is shorter due to later onset of filling (longer IVRT). As the LV relaxes more slowly, the atrial-to-LV pressure gradient is smaller in early diastole and the velocity of filling is lower and filling extends into diastasis. As early diastolic filling is reduced, there is enhanced dependence on atrial contraction for filling. Tachycardia (which shortens diastasis) and loss of atrial contraction (atrial fibrillation) severely impair filling and mandate the need for elevation of left atrial pressures to restore filling and maintain cardiac output. |
There are many extracardiac and cardiac mechanisms that affect load, uniformity, and myofiber inactivation and that may mediate impaired relaxation in HF, and they have been recently reviewed in detail (47).
Increased diastolic stiffness (decreased compliance) is present in patients with HFnlEF and contributes to elevated filling pressure at rest or with exercise. Measurement of diastolic stiffness is considerably more difficult than characterization of relaxation. Ideally, LV pressure and volume are measured during acute alteration in preload to define the end-diastolic pressure-volume relationship over a range of preloads—the multiple-beat method (Figs. 7-4 and 7-5). Alternatively, pressure and volume can be measured over a single beat and the diastolic portion of the pressure-volume curve can be fit to an exponential relationship and used to estimate stiffness.
This single-beat method is subject to two important limitations. First, as previously outlined, the end-diastolic pressure-volume relationship is curvilinear, and operant stiffness measured at increased volume will yield a higher stiffness estimate than when the single beat is taken at lower volumes (see discussion of the third theory, HFnlEF Is Due to Ventricular Remodeling Characterized by LV Dilatation, later). Thus, it could be argued that increased stiffness measured by the single-beat method in HFnlEF could be due to intrinsic diastolic dysfunction or volume overload and LV dilation. This potential limitation can be addressed by carefully documenting that the volumes measured in HFnlEF are normal for age. As values for LV volume are dependent on age, sex, and the method used to measure it, this can be difficult.
The second limitation is due to the influence of impaired relaxation on the contour of the single-beat diastolic pressure-volume relationship. In the normal ventricle, relaxation is brisk and the LV minimum pressure occurs early in diastole, yielding a exponential contour to the diastolic pressure-volume relationship over a single beat. Here, only the passive stiffness of the ventricle influences the pressure-volume relationship in mid- to late-diastole, and the data fit an exponential relationship that allows calculation of a stiffness constant:
P = α × eβ × V
where β is the stiffness constant and α is a curve-fitting constant. However, when relaxation is markedly impaired, relaxation may continue into mid- and even late-diastole under some conditions (5,25); the LV minimum pressure occurs later in diastole; and the diastolic pressure-volume relationship over the single beat is not exponential. Thus, diastolic pressures in mid- to late-diastole are influenced by relaxation as well as the passive characteristics of the ventricle. This limitation is best addressed by using preload reduction to define the end-diastolic pressure-volume relationship, avoiding measurements in other portions of diastole that may be influenced by relaxation. Alternatively, methods to correct for the affect of relaxation on diastolic pressures in the single-beat method have been used (5).
Another important limitation to both the single- and multiple-beat methods for assessment of diastolic stiffness involves the desire to express the position and contour of the exponential diastolic pressure-volume relationship in a single number. In the multiple-beat method, the end-diastolic pressure-volume data points during preload reduction are fit to one of two frequently used exponential equations.
In these equations relating end-diastolic pressure (EDP) and end-diastolic volume (EDV), a curve-fitting constant (α) and a coefficient of stiffness (β) are calculated and the stiffness constant is used to characterize the stiffness of the ventricle. In the second equation, a pressure offset is also calculated (P0) which allows for quantification of a shift in the position of the end-diastolic pressure-volume relationship with or without an accompanying change in the steepness of the relationship. However, as the pressure-volume data acquired in any experiment are not perfectly monoexponential,
fitting the data to these equations can often yield very different values for α and β, even when the curves appear very similar. Both α and β convey information regarding the stiffness of the ventricle, so if β was not increased but α was, one could not conclude that stiffness was normal. Furthermore, this fact can yield considerable variability in group averages for α and β, making it difficult to detect changes.
fitting the data to these equations can often yield very different values for α and β, even when the curves appear very similar. Both α and β convey information regarding the stiffness of the ventricle, so if β was not increased but α was, one could not conclude that stiffness was normal. Furthermore, this fact can yield considerable variability in group averages for α and β, making it difficult to detect changes.
To overcome this limitation, one can use a linear equation to characterize the pressure-volume relationship but the end-diastolic pressure-volume data do not fit a linear equation well. More recently, investigators have recommended converting the exponential pressure volume relationship to a linear one by plotting In EDP versus EDV. This yields a linear relationship:
lnEDP = lnα + β × EDV
One can then compare the slope of this relationship (β) controlling for concomitant changes in the offset (lnα) and for the patient group in multiple linear regression analysis (19). The theoretical value of this method is considerable and deserves further study.
A final limitation to either method of characterizing stiffness with pressure-volume data is the considerable difficulty in accurately and instantaneously measuring volume along with pressure, particularly in clinical studies. While conductance catheter, echocardiography, contrast ventriculography, and radionuclide angiography have been used in clinical studies, all these methods have significant limitations when applied to a clinical population.
These difficulties have limited the characterization of ventricular diastolic stiffness in HFnlEF to only a few studies. Zile et al. used the single-beat method, measuring volumes with echocardiography and correcting for the influence of impaired relaxation on the pressure-volume contour. The patients with HFnlEF (n = 47) were relatively young and predominately male but had a firm diagnosis of HFnlEF (5,38). Compared to age-matched controls without cardiovascular disease, the stiffness coefficient was significantly higher in patients with HFnlEF (Fig. 7-9). These
investigators concluded that both impaired relaxation and increased diastolic stiffness were present in patients with HFnlEF and that these abnormalities in diastolic function were key to the pathogenesis of the syndrome.
investigators concluded that both impaired relaxation and increased diastolic stiffness were present in patients with HFnlEF and that these abnormalities in diastolic function were key to the pathogenesis of the syndrome.
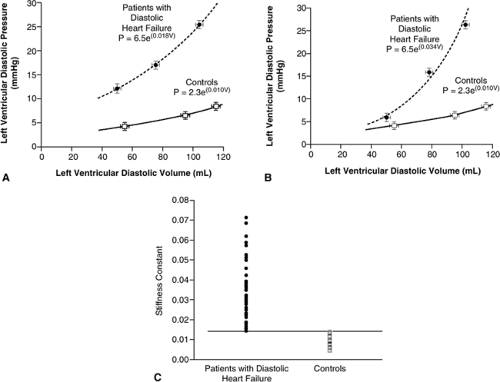 Figure 7-9 Diastolic pressure-volume relation in patients with diastolic heart failure and in controls. A shows measured values for the minimal left ventricular pressure and B shows values for the minimal left ventricular pressure, corrected for slow relaxation. The exponential value in the equation for pressure (P) is the stiffness constant. The data in both panels indicate that there was a significant increase in the passive stiffness of the left ventricle in the patients with HFnlEF (diastolic heart failure). V denotes volume, and the l-bars represent the standard error. C shows the individual corrected stiffness constants in patients with HFnlEF (Diastolic Heart Failure) and in Controls. The stiffness constant was increased in all the patients with HFnlEF. (From Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure—abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med. 2004;350(19):1953–1959 .) |
Consistent with this concept, Liu et al. assessed diastolic stiffness invasively but fit end-diastolic pressure-volume to a linear relationship and found significant increases in diastolic stiffness in 10 patients with hypertrophy and normal EF (seven with HFnlEF) as compared to eight somewhat younger controls (48). Finally, Borbely et al. characterized chamber stiffness and, on biopsy specimens, myocardial structure, sarcomere protein composition, and myocyte stiffness in a group of 12 HFnlEF patients (five with previous cardiac transplant and all with a history of hypertension) and eight controls (six with previous cardiac transplant) (49). HFnlEF patients had increased diastolic stiffness and higher diastolic pressures at comparable indexed LV volumes consistent with increased myocardial diastolic stiffness and reduced chamber distensibility. The HFnlEF patients had more fibrosis than controls and the degree of fibrosis did correlate with LV diastolic pressures. This study showed increased passive diastolic myocyte stiffness that was unassociated with apparent changes in sarcomeric protein composition, although isoform switches in titin were not investigated. These investigators concluded that both the increased fibrosis and increased passive myocyte stiffness contribute to the increased chamber stiffness observed in vivo, with extracellular matrix contributing more as filling pressures (and presumably sarcomere length) increase. The potential contribution of collagen modification by cross-linking related to the presence of advanced glycation end-products (AGE) was not explored and could contribute to an imperfect correlation between collagen content and chamber stiffness. While this study supports a role for diastolic stiffness in HFnlEF, the special nature of the patient population must be recognized.
In contrast, Kawaguchi et al. used the conductance catheter and an inferior vena cava balloon to measure the pressure-volume relationship with the multiple-beat method during preload reduction in 10 HFnlEF patients and nine healthy, age-matched controls without hypertension or symptoms of HF (29). These patients were studied at a referral center and, while they were predominantly female, their mean age was 60 years—a value much lower than the mean age observed in most population-based studies of HFnlEF. In this study, assessment of the stiffness coefficient (β) by the single-beat method showed increased stiffness, and an increase in Po, indicating an increased slope and an upward shift in the diastolic pressure-volume relationship (Fig. 7-10). However, when the multiple-beat method was used, there was still an upward shift in the position of the curve but the diastolic stiffness coefficient was not different in controls and HFnlEF patients. The HFnlEF patients had increased vascular stiffness (Ea), markedly increased ventricular systolic stiffness (Ees), and an exaggerated systolic pressure response to exercise. These investigators concluded that HFnlEF is not solely due to resting diastolic dysfunction but that vascular and ventricular systolic stiffening could limit cardiovascular reserve and exacerbate diastolic dysfunction during exercise, thus contributing to the pathophysiology of HFnlEF. That diastolic stiffness was not increased in the HFnlEF patients is somewhat surprising when taking the concept of time varying elastance into consideration. This concept suggests that if systolic stiffness is increased, diastolic stiffness will be as well, a concept previously confirmed in clinical studies showing concomitant age-related increases in arterial, ventricular systolic, and ventricular diastolic stiffness (50). The large variability in β observed in HFnlEF (range ≈ 0.01 to 0.05 mm Hg/mL) may be related to the limitations in calculating β discussed above previously, and this variability may prevent demonstration of differences in β in small groups. The values for α were not reported.
While most discussion has centered on ventricular compliance in HFnlEF, atrial compliance is a potentially important mediator of mean atrial and, thus, mean pulmonary venous pressures. This, in turn, can contribute to pulmonary congestion. It is important to remember that it is mean left atrial pressures, not simply ventricular end-diastolic pressures, that mediate symptoms of HF (44,45). Thus, early- and mid-diastolic as well as end-diastolic pressures in the ventricular and systolic atrial pressures (atrial V wave) are important contributors to mean left atrial pressures, namely, the resistance to filling that the pulmonary venous system faces. Indeed, an often-forgotten hemodynamic hallmark of restrictive cardiomyopathy is the presence of large V waves in the atrial wave pressure waveform in the absence of mitral regurgitation. This atrial systolic pressure wave can exceed 50 mm Hg and reflects reduced atrial compliance with increases in atrial pressures during the ventricular systolic/atrial diastolic phase of atrial filling. The Doppler correlate of this is the reduction in systolic forward flow in the pulmonary veins seen in advanced diastolic dysfunction.
Thus, although studies are few there is evidence that impaired relaxation and increased diastolic stiffness are present at rest and contribute to the pathophysiology of HFnlEF. Furthermore, even if resting diastolic dysfunction is not markedly perturbed, exercise or hypertensive episodes may lead to exaggerated impairment in relaxation or stiffness in other patients with HFnlEF. Those mechanisms that might contribute to increased LV diastolic stiffness include factors which influence ventricular interaction and extrinsic forces, the extracellular cardiac matrix, the myocyte, and the myofibrils themselves. These postulated mechanisms have been reviewed recently (47).
HfnlEF Is Due to Increased Vascular and Ventricular Systolic Stiffness Associated with Enhanced Sensitivity to Volume and Load/Stress-Induced Diastolic Dysfunction
Previous studies using a variety of indexes have established that large-artery stiffness increases with age (51,52,53), is higher in women (51,54,55,56), and increases with age even in the absence of vascular disease or risk factors (51,52,57). Arterial elastance (Ea) is a valuable means to index net ventricular afterload, which combines mean (systemic vascular resistance) and oscillatory (large-artery stiffness) components of the arterial load (58). Consistent with age-associated increases in other measures of large-artery stiffness,
age-related changes in Ea have also been demonstrated (50). Multiple mechanisms have been proposed to explain age-dependent vascular stiffening, including alterations in endothelial function, structural protein composition, collagen cross-linking, geometric changes, and neurohumoral signaling (59). The cause of gender differences in vascular stiffening is unclear, although most studies indicate that this is not simply a matter of differences in body size and vasculature length (51,55). Hearts coupled to a stiffer vascular system are subjected to higher systolic stresses as well as wider pulse pressures that can adversely influence the regulation of coronary flow (50,51,60,61) and contribute to microvasculature and end-organ damage (51). In order to maintain optimal interaction with the arterial system, the LV itself must develop greater systolic stiffness (50,62,63,64).
age-related changes in Ea have also been demonstrated (50). Multiple mechanisms have been proposed to explain age-dependent vascular stiffening, including alterations in endothelial function, structural protein composition, collagen cross-linking, geometric changes, and neurohumoral signaling (59). The cause of gender differences in vascular stiffening is unclear, although most studies indicate that this is not simply a matter of differences in body size and vasculature length (51,55). Hearts coupled to a stiffer vascular system are subjected to higher systolic stresses as well as wider pulse pressures that can adversely influence the regulation of coronary flow (50,51,60,61) and contribute to microvasculature and end-organ damage (51). In order to maintain optimal interaction with the arterial system, the LV itself must develop greater systolic stiffness (50,62,63,64).
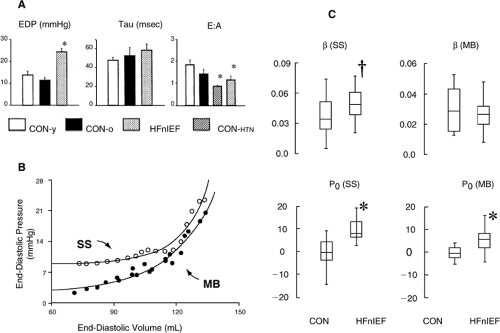 Figure 7-10 A study comparing ventricular function in young healthy persons (CON-y), older non-hypertensive persons (CON-o), patients with HFnlEF, and older hypertensive persons without HF (CON-HTN). A. Mean group data for end-diastolic pressure (EDP), relaxation time constant (Tau), and early/late filling ratio (E:A). B. Example of diastolic pressure volume data and fits from resting steady-state beat (SS) and multibeat analysis after inferior vena cava obstruction (MB). Steady-state data were typically shifted upward and were more abruptly nonlinear due to pressure elevation in the early filling phase. C. Summary data for end-diastolic pressure-volume relation exponential fits from steady-state and multibeat analyses. The stiffness coefficient and the pressure intercept were both elevated in SS analysis, while only the pressure intercept was elevated in MB analysis. Data for all normotensive controls were very similar and were therefore combined (CON). (From Kawaguchi M, Hay I, Fetics B, Kass DA. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction: implications for systolic and diastolic reserve limitations. Circulation 2003;107(5):714–720 .) A. *p <0.001 versus young (CON-y) and age-matched (CON-o) normotensive controls. C. *p <0.001; †p <0.02. |
Importantly, age-associated increases in Ea were associated with increased systolic (Ees) and diastolic LV stiffness (Ed) in a prior invasive study which measured Ees and Ed with invasive pressure volume analysis in individuals of varying age (50) (Fig. 7-11). In this study, gender differences were not explored. However, a population-based study where Ea, Ees, and Ed were measured noninvasively demonstrated age-associated increases in Ea, Ees, and Ed and showed that these indexes were all higher in women than in men, regardless of age (65).
The chronic changes in LV systolic (and potentially diastolic) stiffness that couple with changes in vascular stiffness may represent cardiovascular adaptations to maintain optimal ventricular-vascular matching and, thus, optimal cardiac performance. Whether this is mediated by increases in inotropic state or by structural remodeling such as hypertrophy, remodeling (smaller LV cavity dimensions), or fibrosis (66) is unclear, but altered chamber geometry with age likely plays a role. While concentric hypertrophy or concentric remodeling in response to hypertension or the increases in large-artery stiffness may promote smaller (and stiffer) ventricles in elderly persons,
the possibility that age-related decreases in LV volume are related to inactivity has been suggested (67).
the possibility that age-related decreases in LV volume are related to inactivity has been suggested (67).
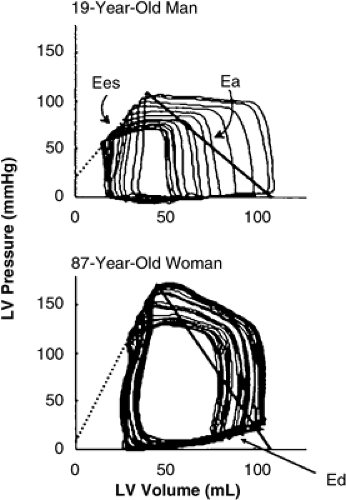 Figure 7-11 Pressure-volume curves obtained with a conductance catheter during acute reduction in preload. An example from a young (top) man and an elderly (bottom) woman are shown. End-systolic (Ees), end-diastolic (Ed), and effective arterial (Ea) elastances are elevated in the elderly woman, indicating cardiovascular stiffening. Grouped data from 57 adults confirm that all three parameters were related to age in otherwise healthy individuals. (From Chen C-H, Nakayama M, Nevo E, et al. Coupled systolic-ventricular and vascular stiffening with age: implications for pressure regulation and cardiac reserve in the elderly. J Am Coll Cardiol. 1998;32(5):1221–1227 .) |
While vascular-ventricular coupling helps to maintain stroke volume and mechanical efficiency, increases in Ees may have adverse effects. Increases in Ees result in increased sensitivity of systolic pressure to changes in volume (50). Thus, volume overload in such individuals could be associated with greater increases in systolic blood pressure and with greater increases in diastolic LV pressures. Together, increases in arterial and systolic stiffness promote load-induced impairment in LV relaxation, as previously outlined (29). Thus, age-related vascular-ventricular stiffening could contribute to increases in LV systolic blood pressure, tau, and LV diastolic pressures with exercise and could predispose to HFnlEF. As previously discussed, the recent study by Kawaguchi et al. in HFnlEF patients and age-matched controls demonstrated increased arterial and LV systolic stiffness and exaggerated increases in tau and LV diastolic pressures with exercise in HFnlEF patients as compared to normals (Fig. 7-3). While this study suggested that increases in diastolic stiffness need not be present in HFnlEF patients with increased arterial and LV systolic stiffness, it is reasonable to expect that such increases may be present in some or many patients.
The role of vascular and ventricular systolic stiffening in mediating symptoms in large numbers of HFnlEF patients has not been defined but is certainly a plausible explanation for many of the features of HFnlEF. Such age- and cardiovascular disease-associated changes in arterial and ventricular properties also are consistent with the epidemiology of HFnlEF where dramatic increases in the prevalence of HFnlEF occur with age and particularly in women (Fig. 7-12) (68). As previously discussed at length, these
mechanisms do not exclude concomitant changes in resting diastolic function, either impaired relaxation or increased diastolic stiffness in some or in many persons with HFnlEF. These mechanisms do, however, provide a paradigm for understanding exercise- or other stress-induced symptoms in patients in whom resting diastolic function does not seem markedly perturbed.
mechanisms do not exclude concomitant changes in resting diastolic function, either impaired relaxation or increased diastolic stiffness in some or in many persons with HFnlEF. These mechanisms do, however, provide a paradigm for understanding exercise- or other stress-induced symptoms in patients in whom resting diastolic function does not seem markedly perturbed.
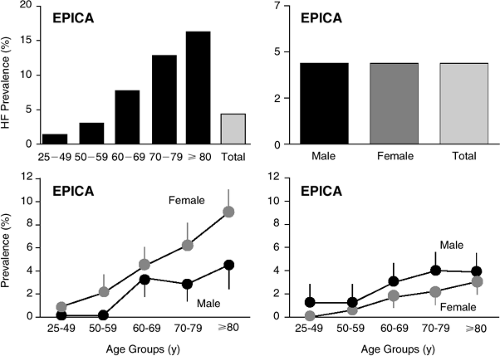 Figure 7-12 Data from a large epidemiology study in Portugal demonstrate the well-established increase in clinical HF prevalence with age (top left). In this study, the prevalence of HF was similar in men and women (top right). HFnlEF prevalence (bottom left) increased much more sharply with age than did the prevalence of HF↓EF (bottom right) and this was especially apparent in women. (From Ceia F, Fonseca C, Mota T, et al. Prevalence of chronic heart failure in Southwestern Europe: the EPICA study. Eur J Heart Fail. 2002;4(4):531–539 .) |
HFnlEF Is Due to Ventricular Remodeling Characterized by LV Dilation with Volume-Dependent Elevation of Filling Pressures Unassociated with Significantly Altered Diastolic Stiffness or Impaired Relaxation
It has been assumed that patients with HFnlEF have normal or reduced LV volumes. This assumption is based on many studies which have reported, on average, normal LV cavity dimensions or volumes in patients with HFnlEF (9,13,69,70,71,72,73,74,75,76,77). In these types of studies where only grouped data are presented as a mean, the standard deviation of the dimension or volume measurements is wide and the potential for subgroups of patients to have enlarged, normal, and reduced cavity dimensions is almost certain.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


