Michael R. Zile, William C. Little Patients with heart failure can be divided into those with (1) heart failure with a reduced ejection fraction (HFrEF) and (2) heart failure with a preserved ejection fraction (HFpEF). All of these patients, regardless of ejection fraction status (EF value), have the clinical syndrome of heart failure. In addition, many features are similar across the EF spectrum, including abnormal left ventricular (LV) filling dynamics, elevated LV diastolic pressure, LV systolic and diastolic dysfunction, neurohormonal activation, impaired exercise tolerance, frequent hospitalization, and reduced survival.1–4 Patients with HFpEF have a devastating 5-year mortality rate (approaching 60%), costly morbidity (6-month hospitalization rate of 50%), and debilitating symptoms (maximum myocardial oxygen consumption [MVo2] averaging 14 mL/g/min).5,6 Clear differences also are recognized between HFpEF and HFrEF. Compared with those with a reduced EF, patients with preserved EF are older and more likely to be female; however, HFpEF occurs in both men and women throughout the 5th to the 9th decades of life.7 The most common antecedent disease leading to HFpEF is systolic hypertension, which is present in more than 85% of patients, whereas ischemic heart disease is much less common than in HFrEF.7 Differences in cardiovascular structure and function between HFpEF and HFrEF also are well recognized.4,8–12 Patients with HFpEF have normal LV end-diastolic volume and normal (or near-normal) EF and stroke volume and commonly exhibit concentric remodeling of either LV chamber and/or cardiomyocytes. Finally, differences also are evident in the effects of pharmacologic treatment in patients with HFrEF versus HFpEF. Standard heart failure therapy shown to be effective in HFrEF has not been found to reduce morbidity or mortality associated with HFpEF, leaving a substantial area of unmet need.13 This chapter summarizes the current understanding of the clinical, prognostic, pathophysiologic, and therapeutic information about patients with HFpEF and suggests where future advances are likely to occur. A variety of terms have been used to describe patients with what is now called heart failure with a preserved ejection fraction (HFpEF). These terms include heart failure with a normal EF, heart failure with normal systolic function, diastolic heart failure, and diastolic dysfunction heart failure. Heart failure guidelines, recent publications and this chapter use the term HFpEF. The mean EF in normal populations depends somewhat on the method used to measure it but generally is agreed to be above 60%. The lower 95% confidence limit for EF is approximately 55%, whereas an EF greater than 50% often is used as a diagnostic criterion for HFpEF; this term also has been applied to heart failure in some patients with EFs outside the normal range. For example, some randomized controlled trials (RCTs) have included patients with EFs greater than 35%, 40%, or 45%. Therefore, because the term HFpEF has been applied to this broader spectrum of patients with heart failure, the term “heart failure with a normal EF” is less commonly used. Although it is clear that patients with an EF below 50% have abnormalities in systolic function, recent studies have shown that even patients with an EF above 50% may have midwall and/or longitudinal systolic dysfunction. Therefore the term “heart failure with normal systolic function” is not an accurate description even for patients with heart failure and EF above 50%. Because patients with the clinical syndrome of HFpEF have abnormalities in LV diastolic function, systolic function, and vascular properties; the terms “diastolic heart failure” and “diastolic dysfunction heart failure,” which single out the abnormalities in diastole, are now used less commonly. The term diastolic dysfunction refers to abnormalities in LV filling secondary to altered compliance, relaxation, and/or recoil. Abnormalities in diastolic function can occur in the presence or absence of a clinical syndrome of heart failure and with normal or abnormal systolic function. Whereas diastolic dysfunction describes abnormal LV performance, HFpEF describes a clinical syndrome of heart failure. A substantial proportion (more than 50% in many studies) of patients who are diagnosed or hospitalized with heart failure have HFpEF.14,15 This observation is consistent among different racial groups, geographic regions, and hospital- and community-based studies. The prevalence of HFpEF increases dramatically with age, and this heart failure syndrome is much more common in women than in men at any age (Fig. 27-1). The prevalence of HFpEF appears to be increasing, perhaps as a function of the aging population and increased recognition of the clinical entity. The distribution of EF across unselected populations of patients with heart failure is bimodal, with peaks centered on 35% and 55%16 (Fig. 27-2). These data further emphasize that HFpEF is an important cause of the heart failure syndrome. The 5-year survival rate for all patients with heart failure, regardless of EF, is less than 50%. Although survival has improved over time for patients with HFrEF, it has not changed for patients with HFpEF (Fig. 27-3A, B).14 Some epidemiologic studies have found that all-cause mortality for HFpEF is similar to that for HFrEF; other epidemiologic studies and RCTs suggest that all-cause mortality is somewhat lower in HFpEF than in HFrEF (see Chapter 25). For example, three RCTs have enrolled both patients with HFpEF and those with HFrEF, allowed direct comparisons, and demonstrated a lower mortality rate in HFpEF versus HFrEF.17 Taken together, data from epidemiologic studies of HFpEF find that the annual mortality is approximately 10%, but RCTs in patients with HFpEF suggest that the annual mortality is about 5%. This apparent difference may be due to the exclusion of patients with the comorbid conditions from the RCTs. However, the mortality rates found in patients with HFpEF are not solely due to the comorbid diseases. In the RCTs, patients with HFpEF, who have antecedent and comorbid factors such as hypertension, coronary artery disease. and diabetes mellitus, were found to have more than twice the mortality rate of patients with hypertension, coronary artery disease, or diabetes who do not have HFpEF17 (Fig. 27-3C, D). Most (>70%) of the deaths in patients with HFpEF are cardiovascular in nature, with 20% due to heart failure and 35% due to sudden death18 (Table 27-1). This distribution of modes of cardiovascular death is similar to that for HFrEF. The incidence of noncardiovascular deaths is significantly higher for HFpEF (30%) than for HFrEF (15%), reflecting the higher age and increased comorbidity in patients with HFpEF. Among patients with HFpEF, morbidity rates are comparable to those with HFrEF; heart failure–related hospital readmission rates approximate 50% at 6 months for both HFrEF and HFpEF. The lifetime burden for all-cause hospitalization is high and nearly equivalent for patients with HFpEF and those with HFrEF. All patients with heart failure (both HFpEF and HFrEF) have many comorbid conditions that influence both morbidity and mortality.19 Like mortality, the heart failure hospitalization rates in patients with HFpEF are not based solely on the presence of antecedent or comorbid conditions themselves. Data from RCTs have shown that the heart failure hospitalization rates in patients with HFpEF (who have antecedent and comorbid factors such as hypertension, coronary artery disease, and diabetes mellitus) are more than twice than those in patients with hypertension, coronary artery disease, or diabetes mellitus who do not have HFpEF (see Fig. 27-3D).17 Rates of progressive functional decline after hospital admission for heart failure also are similar in patients with HFpEF and in those with HFrEF. In addition to heart failure–related hospitalizations, abnormalities in exercise tolerance, MVo2, and quality-of-life assessment are similar for HFrEF and HFpEF. The pathophysiologic mechanisms that cause the development of HFpEF are reflected in changes in LV relaxation and filling, LV structural remodeling and altered geometry, and changes in LV and vascular compliance (Table 27-2). TABLE 27-2 Mechanisms/Factors Contributing to the Pathophysiology of Heart Failure with Preserved Ejection Fraction SPARC = secreted protein, acidic and rich in cysteine [osteonectin]. Normal diastolic function (see Chapter 21) allows the ventricle to fill adequately during rest and exercise, without an abnormal increase in left atrial (LA) pressure. The phases of diastole are isovolumic pressure decline and filling. The filling phase is divided into early rapid filling, diastasis, and atrial systole. Early rapid filling contributes 70% to 80% of LV filling in normal individuals. This contribution diminishes with age and various disease states. Early diastolic filling is driven by the LA-to-LV pressure gradient, which is dependent on a complex interplay of factors—myocardial relaxation, LV elastic recoil, LV diastolic stiffness, LA pressures, ventricular interaction, pericardial constraint, pulmonary vein properties, and mitral orifice area. Diastasis occurs in mid-diastole, when the LA and LV pressures usually are almost equal. It contributes less than 5% of the LV filling, and its duration shortens with tachycardia. In normal subjects, atrial systole contributes 15% to 25% of LV diastolic filling without raising the mean LA pressure. This contribution depends on the PR interval, atrial inotropic state, atrial preload, atrial afterload, autonomic tone, and heart rate. LV relaxation is an active, energy-dependent process that begins with the decay of force-generating capacity, follows the completion of the ejection phase of systole, and continues through isovolumic pressure decline and the rapid filling phase. Filling is dependent both on active relaxation and on the recoil/suction that results from the release of potential energy stored during systole by contraction. Thus blood is effectively “pulled” into the left ventricle.23 In normal hearts, over a range of normal heart rates, relaxation and recoil are adequate to allow LA pressures to remain normal. In addition, catecholamine-induced enhancement of relaxation and recoil during exercise lowers LV pressures in early diastole, thereby increasing the LA-to-LV pressure gradient without increasing LA pressures as well as enhancing filling during exercise. By contrast, in patients with HFpEF, relaxation and recoil are abnormal at rest and are not enhanced during increased HR or exercise. As a result, filling can be maintained only by increased LA pressure; blood must be “pushed” into the left ventricle. Isovolumic Pressure Decline. The time course of isovolumetric pressure decline has been quantitatively described by the peak rate of pressure fall (dP/dtmin) and the time constant τ (tau) of the exponential fall in LV isovolumetric pressure. Each of these requires that LV pressure be measured using a micromanometer-tipped catheter. dP/dtmin measures the rate of pressure decline at a single point in time, is strongly influenced by the LV pressure at the time of aortic valve closure, and therefore, like all indices of diastolic function, is afterload-dependent. Patients with HFpEF have a larger dP/dtmin, signifying that relaxation rate is decreased. The time constant τ describes the rate of LV pressure decline throughout isovolumic relaxation. Pressure (P) and time (t) data during the period from end-systole (aortic valve closure) to the onset of LV filling (mitral valve opening) are fit to an exponential equation such as the following: LV pressure = P0e−t/τ, where P0 is LV pressure at end-ejection and τ is the exponential time constant. The larger the value of τ, the longer it takes for the LV pressure to fall and the more impaired is relaxation. A normal value for τ is less than 40 milliseconds in most age groups, suggesting that relaxation is nearly complete by 3.5 × τ (less than 140 milliseconds). The isovolumic relaxation time (IVRT) also can be estimated by echo techniques as the time between aortic valve closure and mitral valve opening. Although less precise than τ, IVRT is useful in the noninvasive assessment of diastolic properties. However, IVRT depends not only on the rate of LV relaxation but also on the aortic pressure at the time of aortic valve closure and the LA pressure at mitral valve opening. Thus IVRT can be increased by an elevation of aortic pressure or decreased by an increase in LA pressure. The time course of LV pressure decline during isovolumetric relaxation can also be characterized using noninvasive Doppler measurement of the velocity of a regurgitant jet across the mitral valve. In this method, the modified Bernoulli equation is used to approximate LV pressure during isovolumetric relaxation, allowing calculation of the maximum rate of LV pressure decline and the exponential time constant. Recoil and Left Ventricular Filling. During systole, potential energy is stored in the elastic elements of the cardiomyocytes and extracellular matrix (ECM).23 The elastic elements are compressed and twisted during systolic contraction. During relaxation, this potential energy is released as the elastic elements recoil and return to their original length and orientation. Recoil causes LV pressure to fall rapidly during isovolumetric relaxation. Furthermore, for the first 30 to 40 milliseconds after mitral valve opening, the relaxation of LV wall tension normally is rapid enough to cause LV pressure to continue to decline despite an increase in LV volume. This fall in LV pressure produces an early diastolic pressure gradient from the LA that extends to the LV apex (see Because the LV apex remains fixed during the cardiac cycle, the mitral annular velocity provides a measure of long-axis lengthening rate.28 Under normal conditions, peak early diastolic mitral annular velocity (e′) occurs coincidentally with or before the mitral E (see Fig. e27-1A, B).29,30 This is a manifestation of the symmetric expansion of the left ventricle in early diastole as blood moves rapidly to the LV apex in response to a progressive pressure gradient from the left atrium to the LV apex. In addition, the rapid recoil of the mitral annulus and valve into the left atrium early in diastole relocates blood from the left atrium into the left ventricle. Under normal circumstances, both E and e′ respond to changes in the LA-to-LV pressure gradient. For example, both E and e′ normally increase in response to increased volume load and exercise.30–32 LV relaxation is under the control of multiple factors that include hemodynamic load (early diastolic load and afterload), myofiber inactivation (see discussion of cellular determinants further on), and the uniformity of the distribution of load and inactivation in space and time (dyssynchrony, dyssynergy, Treppe). Each of these determinants may affect indices of diastolic relaxation, recoil and filling. Both isovolumic pressure decline and early filling are affected by afterload (LV systolic stress). An increase in LV systolic stress results in a delay in and slowed rate of pressure decline and early filling. Increases in systolic load may have different effects, depending on when the load is imposed during systole. Increases in LV pressure late in systole hasten the onset of LV relaxation, but relaxation occurs at a slower rate (increased τ). Increases in LV pressure late in systole occur with aging because of age-related vascular stiffening, which alters the timing of reflected pressure wave in the vascular tree so that the reflected wave arrives in late systole rather than diastole. In clinical practice, an acute increase in blood pressure either at rest or during exercise will impair ejection, slow pressure decline, prolong time to complete relaxation, and reduce recoil. These changes in relaxation decrease the LA-to-LV gradient, decrease early filling, and result in increased LV diastolic and LA pressure. In addition, the load present at the time of mitral valve opening (LA-to-LV gradient, i.e., early diastolic load) affects early LV filling. Synchrony (timing of relaxation of the different myocardial segments) and synergy (extent to which myocardial segments relax) will enhance LV relaxation, whereas dyssynchrony or dyssynergy (e.g., caused by infarction, ischemia, asymmetry of hypertrophy, or conduction abnormalities) will impair global LV relaxation. Dyssynchrony, measured using a variety of echocardiographic measurements, may be present in patients with HFpEF, particularly those with left bundle branch block (LBBB) or right ventricular (RV) pacing. Whether or not treatment aimed at resynchronization (i.e., cardiac resynchronization therapy [CRT]) will effect clinical improvement in patients with HFpEF is currently under investigation. Myofiber inactivation refers to the many cellular processes (Chapter 21) that ultimately influence the process by which the left ventricle, its constitutive cardiomyocytes and individual sarcomeres return to a normal end-diastolic length with minimum cross-bridge cycling and low force generation. To accomplish this state of complete relaxation requires (1) calcium resequestration into the sarcoplasmic reticulum, followed by calcium extrusion into the extracellular space; (2) availability of sufficient ATP; (3) normal myofilament function; and (4) normal elastic properties of the cardiomyocyte and the ECM. Additional material on this topic is presented in an Impaired relaxation is present in HFpEF and contributes to the development of elevated LA pressure at rest. The rate of relaxation is further impaired during exercise and hemodynamic stress. Any factor that shortens the diastolic filling period (prolonged contraction or long PR interval) will enhance the effect of impaired relaxation on LV diastolic pressures during filling and thus affect the mean LA pressure needed to fill the left ventricle. Whether therapies to enhance relaxation directly and specifically can be developed, and whether such therapies will relieve symptoms, remains an area of active investigation. The passive characteristics of the left ventricle during diastole can be described by the passive diastolic pressure-volume relationship (DPVR).23 Optimally, this relationship should be constructed from points that are obtained after relaxation is complete and at slow filling rates, so that viscous effects are not present. In practice, this can be approximated using points obtained late in diastole, when relaxation is assumed to be complete, by correcting pressure data affected by incomplete relaxation or by using data from variably loaded beats at end-diastole. The resultant DPVR is nonlinear and can be approximated by an exponential function. LV stiffness is defined as the ratio of LV diastolic pressure and LV diastolic volume (LV dP/dV) at any given LV diastolic volume. LV compliance is the reciprocal of stiffness (LV dV/dP). Because the DPVR can be approximated as an exponential, stiffness will increase as the left ventricle fills to higher LV diastolic volumes; thus as the left ventricle fills, it becomes stiffer. LV diastolic distensibility is defined as the end-diastolic pressure required to distend the left ventricle to an end-diastolic volume. Patients with HFpEF have reduced distensibility, indicated by a normal or reduced end-diastolic volume and an elevated end-diastolic pressure.2,33 Because the DPVR can be approximated by an exponential function, its position and shape can be described by the constants within an equation such as the following: P = α × eβV, where α and β represent the “stiffness constants.” It should be recognized that β does not indicate stiffness but instead describes how rapidly stiffness increased with increases in volume (β = [dP/dV]/V). The “stiffness constants” derived in this fashion can be used to compare passive diastolic properties in different patients or patient groups. The end-diastolic pressure-versus-volume ratio (instantaneous operative distensibility) also can be used in comparing patients or patient groups. Patients with HFpEF have abnormal DPVRs with elevated β and abnormal distensibility (Fig. 27-4). Patients with heart failure and an increased LV diastolic pressure can be divided into four groups defined by patterns of DPVR, as depicted in Figure 27-5. DPVR in patients with HFrEF typically is characterized by the graphed curve D in the figure, in which eccentric remodeling results in a shift of the DPVR to the right, representing an increase in distensibility. It should be recognized that although the ventricle is more distensible, the LV end-diastolic volume in these patients typically is very large and the end-diastolic stiffness in the operating region is high. DPVR in patients with HFpEF may be characterized by graphed curves A to C. In Figure 27-5C, pericardial constraint causes a parallel upward shift in the DPVR. In patients with HFpEF, when relaxation is markedly prolonged and diastole is abbreviated (as seen in Fig. 27-5A), LV diastolic pressure falls throughout diastole but remains increased. In the most prevalent pattern in HFpEF (depicted in Fig. 27-5B), the DPVR is shifted upward and to the left, indicating reduced distensibility, where LV pressure is increased at any LV volume. Two of the determinants associated with an upward and leftward shift of the DPRV in patients with HFpEF are the presence of LV and cardiomyocyte concentric remodeling and hypertrophy and changes in the material properties of myocardial muscle itself (i.e., myocardial stiffness). Myocardial diastolic stiffness can be determined by assessing the myocardial diastolic LV stress versus. strain relationship. The stress-strain relationship represents the resistance of the myocardium to stretch (increase in length) when subjected to stress (distending force). Calculation of stress requires the use of a geometric model of the LV, and the calculation of strain requires assumption of the unstressed LV volume, which cannot be directly measured in the intact circulation. In addition to these potential theoretical limitations, these calculations require accurate measurements over a wide range of LV pressures, volumes, dimensions, and wall thicknesses. These challenges in determining myocardial stress-strain relationships have limited their clinical application, but they remain important to basic and translational research efforts. LV DPVR is affected by factors that influence the cardiac ECM such as fibrillar collagen, cellular structure and processes at the cardiomyocyte level such as calcium homeostasis and energetics (discussed previously), and myofilament and cytoskeletal proteins such as titin and microtubules.34–36 The ECM consists of fibrillar proteins including collagen type I and type III, elastin, and proteoglycans; basement membrane proteins such as collagen type IV, laminin, and fibronectin; and a large number of bioactive peptides and proteins such as matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), signaling proteins such as transforming growth factor-β (TGF-β), and cytokines (see also Chapter 22). The myocardial collagen network is composed of endomysial fibers surrounding individual myocytes and capillaries; perimysial fibers, which interweave muscle bundles; and epimysial fibers, which form a matrix adjacent to the epicardial and endocardial surfaces. ECM structure is dynamic and regulated by physical, neurohormonal, and inflammatory mediators. These modulate the four steps in collagen homeostasis: collagen synthesis, postsynthetic processing, posttranslational cross-linking, and degradation (see Chapter 22).37–40 The ECM fibrillar collagen content is increased in patients with HFpEF (Fig. 27-6). Experimental studies have shown that acute degradation of collagen fibers by collagenase perfusion or activation of MMPs results in decreased LV stiffness. Animal models have demonstrated that interventions associated with increases or decreases in myocardial fibrosis are associated with increased or decreased LV diastolic stiffness. Thus evidence that the ECM can contribute to diastolic dysfunction by increasing diastolic stiffness or contributing to impairment in relaxation by altering regional loading or uniformity is strong and supports the potential therapeutic strategy of preventing or reducing fibrosis in the therapy of HFpEF. The giant myocardial protein titin spans the Z-lines and serves as a molecular spring that resists distension, thereby contributing to LV stiffness (Chapter 21). A number of factors, including titin isoform switches (to a less compliant N2B isoform) and titin phosphorylation state, affect diastolic stiffness. Such alterations in titin are present in HFpEF contributing to increased LV diastolic stiffness.41,42 Interaction of titin with other signaling molecules and with ion channels also may contribute to diastolic stiffness. The role of alterations in titin and the interactions of titin with the ECM in patients with HFpEF constitute an important area of investigation. In addition to titin, other cardiomyocyte structural proteins and changes in their phosphorylation state may affect diastolic stiffness. These include changes in myosin-binding proteins, microtubules, and others. Measuring the DPVR in large series of patients with HFpEF, particularly in RCTs, is impractical. However, several studies using both invasive measurements and noninvasive estimates of LV stiffness have shown that LV diastolic stiffness is increased in patients with HFpEF as compared with both age-matched control cohorts and patients with hypertensive LV hypertrophy but without heart failure.2,33,43,44 The exact prevalence in either epidemiologic or pathophysiologic studies is not completely defined, but studies to date suggest that the prevalence of increased diastolic stiffness is high in HFpEF. Several studies using implantable hemodynamic monitors have shown that increased LV diastolic pressures (or their equivalents in pulmonary artery diastolic pressure, left atrial pressures) in patients with HFpEF predict an increase in the frequency of subsequent acute decompensated heart failure. The diagnosis of HFpEF requires that the patient have signs and symptoms of heart failure, an EF greater than 50%, and objective evidence of cardiac dysfunction45 (Fig. 27-7). The clinical manifestations of heart failure are similar regardless of the EF. These include reduced exercise tolerance, dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, peripheral edema, and pulmonary congestion apparent on chest radiographs (see also Chapter 23). Although a displaced LV apical impulse and pulsus alternans are presumed to be present only in HFrEF, no clinical features (symptoms, signs, or chest radiography) can be used to reliably distinguish between HFpEF and HFrEF. Thus determination of the EF (usually by echocardiography) is required in patients being evaluated for heart failure. Furthermore, symptoms and signs common in heart failure can have other causes not related to heart failure. For example, exercise intolerance and dyspnea may be due to obesity, pulmonary disease, anemia, or deconditioning. Edema may result from obesity or venous insufficiency. For these reasons, objective demonstration of cardiovascular dysfunction and/or remodeling are necessary to confirm the diagnosis of heart failure. A reduced EF provides this evidence in patients with HFrEF, but in HFpEF the EF is not abnormal (i.e., EF >50%) and the end-diastolic volume is not increased, so an elevation of the biomarker B-type natriuretic peptide (BNP) (or its N-terminal pro form), abnormal LV diastolic function (determined noninvasively or by direct measurement of LV diastolic pressure), or elevated LA volume is required for the diagnosis of HFpEF. Finally, the diagnosis of HFpEF requires the exclusion of noncardiac causes of symptoms and signs. Biomarkers. The best-characterized biomarkers in patients with HFpEF are the natriuretic peptides, BNP and N-terminal proBNP (NT-proBNP). Circulating levels of these proteins are elevated in patients with HFpEF as compared with those in persons without heart failure but are lower than in patients with HFrEF (see also Chapter 23
Heart Failure with a Preserved Ejection Fraction
Overview
Terminology
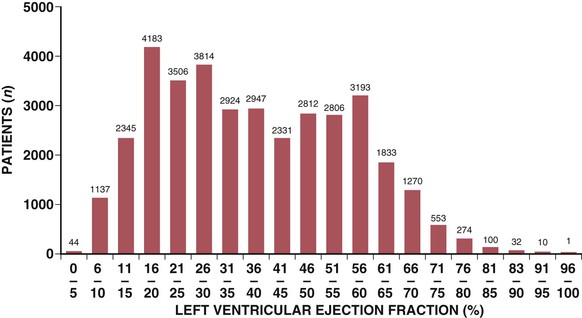
Epidemiology
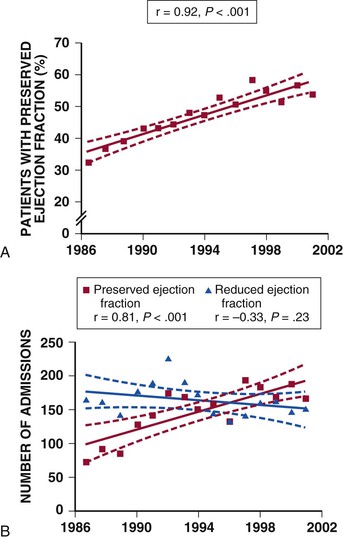
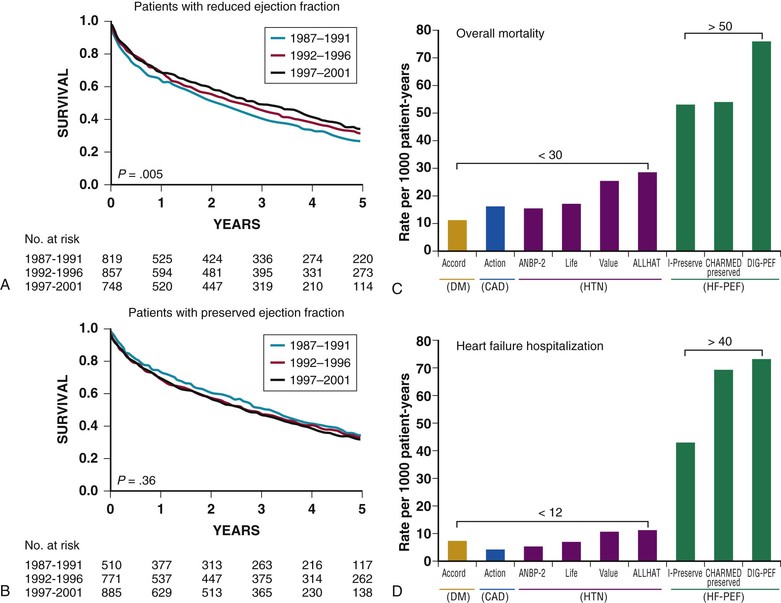
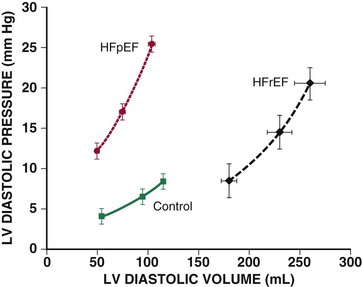
Natural History
Mortality
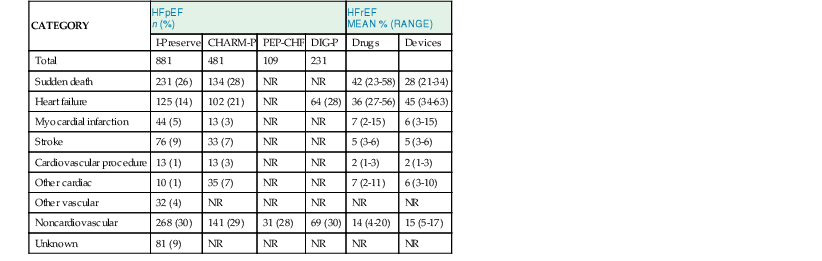
Mode of Death
Morbidity
Pathophysiology
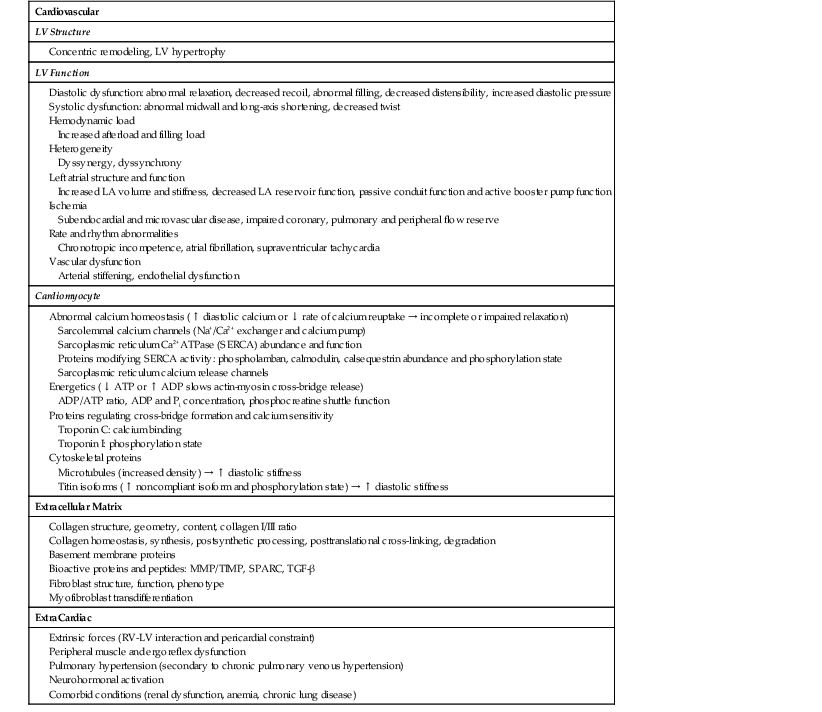
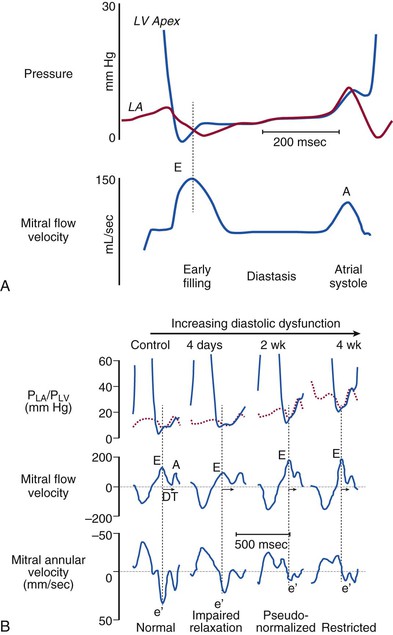
Normal Diastolic Properties
Left Ventricular Relaxation
 Fig. e27-1A). This accelerates blood out of the LA and produces rapid early diastolic flow that quickly propagates to the apex. Because the diastolic intraventricular pressure gradient pulls blood to the apex, it can be considered a measure of LV suction. It is reduced in both experimental models and in patients with ischemia, hypertrophic cardiomyopathy,24 and heart failure including HFpEF.25–27 The intraventricular pressure gradient can be measured noninvasively from the diastolic spatial-temporal velocity map obtained using apical color M-mode echocardiography.
Fig. e27-1A). This accelerates blood out of the LA and produces rapid early diastolic flow that quickly propagates to the apex. Because the diastolic intraventricular pressure gradient pulls blood to the apex, it can be considered a measure of LV suction. It is reduced in both experimental models and in patients with ischemia, hypertrophic cardiomyopathy,24 and heart failure including HFpEF.25–27 The intraventricular pressure gradient can be measured noninvasively from the diastolic spatial-temporal velocity map obtained using apical color M-mode echocardiography.
Determinants of Left Ventricular Relaxation
Hemodynamic Load
Heterogeneity
Cellular Mechanisms
 online supplement for this chapter (Cellular Mechanisms of Myocardial Relaxation).
online supplement for this chapter (Cellular Mechanisms of Myocardial Relaxation).
Prevalence and Prognosis for Abnormal Relaxation
Left Ventricular Diastolic Stiffness, Compliance, and Distensibility
Methods of Measurement
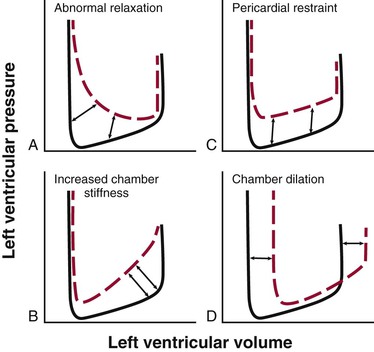
Determinants of Left Ventricular Pressure-versus-Volume Relationship
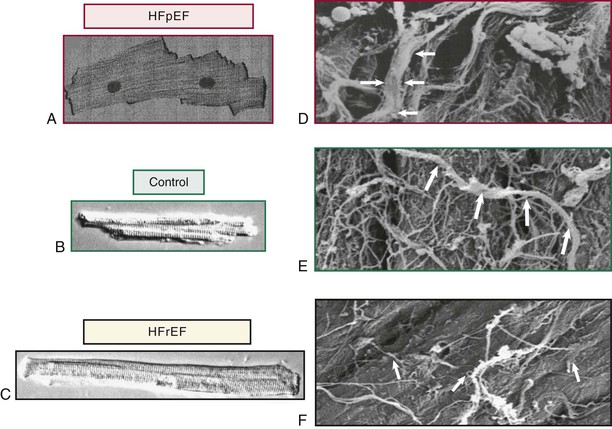
Extracellular Matrix
Myofilament and Extramyofilament Proteins
Prevalence and Prognosis for Decreased Diastolic Distensibility
Clinical Features
Diagnostic Criteria
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Heart Failure with a Preserved Ejection Fraction
27