Heart Failure Sedondary to Coronary Artery Disease
Roger M. Millis Jr.
Mourz Al-Mallah
This chapter reviews current important basic and clinical aspects of heart failure that occur as a consequence of coronary artery disease (CAD). Table 17-1 outlines some common clinical presentations of this problem. Medical management of end-stage ischemic cardiomyopathy and interventions for the complications of acute myocardial infarction (MI) are covered in another chapter; this chapter will concentrate on the pathophysiology and diagnosis of heart failure associated with potentially reversible myocardial ischemia.
In the United States, CAD remains the most common identifiable process underlying heart failure (1). In the ADHERE database, with over 100,000 heart failure admissions, 55% of patients have evidence of CAD (2). Once overt heart failure occurs, the prognosis for patients with established CAD is poor, with an average annual mortality rate of 30% to 40% (3,4,5). In older studies, patients who developed heart failure due to CAD had a significantly worse prognosis than those whose heart failure results from nonischemic pathology. Franciosa et al. followed 182 patients with chronic left heart failure and reported 1- and 2-year mortality rates of 46% and 69%, respectively, in patients with CAD as compared with 23% and 48%, respectively, in those with idiopathic dilated cardiomyopathy (6). Similarly, in a study of 201 patients with advanced heart failure, Likoff et al. found a 6-month mortality rate of 65% in patients with ischemic disease versus 40% in the noncoronary group (7). In a series of 860 patients with known or suspected CAD evaluated at the Mayo Clinic, Chuah et al. reported that a history of heart failure was the most important clinical predictor of cardiac events (hazard 4.26) (8). In a study of 3,787 patients who underwent catheterization at Duke University Medical Center, Bart et al. found significantly higher 5-year mortality rates in ischemic versus nonischemic cardiomyopathy. The 5-year adjusted Kaplan-Meier survival statistic was 0.59 for ischemic and 0.69 for nonischemic patients (p <0.0001) (9) (Fig. 17-1).
This difference persisted despite vasodilator therapy, as demonstrated by the Veterans Administration Cooperative study data shown in Figure 17-2, which compared heart failure patients with coronary and noncoronary causes treated with vasodilators versus the then-conventional therapy or placebo. The impact of today’s state-of-the-art therapy with neurohormonal blockade, antiplatelet agents, revascularization, and arrhythmia control devices on the prognosis of patients with coronary disease and heart failure has not been documented. However, the Duke Cardiovascular Disease Database observational data strongly support the concept that surgical revascularization benefits patients with impaired left ventricular function and CAD (10).
The approach to diagnosis and management of heart failure associated with coronary disease should focus on assessing the possibility that mechanical revascularization with percutaneous transluminal coronary angioplasty (PTCA) or coronary artery bypass grafting (CABG) might relieve ischemia, alleviate symptoms, and improve both cardiac function and clinical outcome. In this setting, a comprehensive approach might include ventricular remodeling, mitral valve repair, epicardial lead placement for resynchronization, and cardiac restraint device placement as well as revascularization, followed by long-term neuro-hormonal blockade, antiplatelet and lipid-lowering therapy, and consideration for implantable cardioverter defibrillator (ICD) placement.
Keeping this clinical strategy in mind, this chapter will first review the pathophysiology of ischemia to clarify how
reduction in coronary blood flow produces hemodynamic dysfunction. A discussion of the role of echocardiography, radionuclide imaging, stress testing, and catheterization laboratory studies in the evaluation of the heart failure patients with known or suspected CAD follows. The brief final section of the chapter offers suggestions for evidence-based decision making and the choice of therapy.
reduction in coronary blood flow produces hemodynamic dysfunction. A discussion of the role of echocardiography, radionuclide imaging, stress testing, and catheterization laboratory studies in the evaluation of the heart failure patients with known or suspected CAD follows. The brief final section of the chapter offers suggestions for evidence-based decision making and the choice of therapy.
Table 17-1 Syndromes of Heart Failure with Coronary Artery Disease | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Physiology of Ischemic Left Ventricular Dysfunction
Coronary Blood Flow and Myocardial Oxygen Demand
Atherosclerosis in the coronary arterial tree manifests as clinical CAD when it produces either reversible or irreversible myocardial oxygen supply—demand imbalance. Understanding heart failure due to CAD requires an appreciation of the precise coupling of coronary blood flow with myocardial oxygen demand and myocardial function. This coupling, normally mediated through the integrated responses of coronary endothelium and vascular smooth muscle (11,12), allows coronary blood flow to increase markedly in response to augmented myocardial metabolic demands. This phenomenon is described as coronary flow reserve.
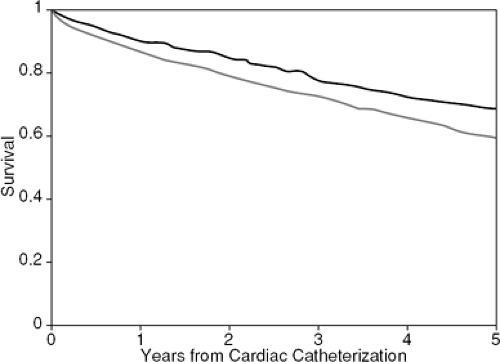 Figure 17-1 Adjusted Kaplan-Meier survival estimates for patients with nonischemic (solid line) and ischemic (dashed line) cardiomyopathy (p <0.0001). (Adapted from Bart BA, Shaw LK, McCants CB, et al. Clinical determinants of mortality in patients with angiographically diagnosed ischemic or non-ischemic cardiomyopathy. J Am Coll Cardiol. 1997;30:1002–1008 .) |
Coronary flow reserve can be measured directly in open-chest laboratory animal experiments using a mechanical snare to constrict the proximal (conduit) vessel and an electromagnetic flow meter to measure distal flow. In this setting, coronary flow at rest and after maximal
coronary resistance vessel vasodilation can be compared while imposing various degrees of proximal epicardial conduit vessel stenosis. Using this technique and intra-coronary injections of radiographic contrast as a vasodilator stimulus, Gould et al. (13) demonstrated that coronary flow with unobstructed vessels increased four- to fivefold with maximal vasodilation. Although resting blood flow did not fall until an 85% proximal stenosis was imposed, the normal four- to fivefold coronary flow reserve was impaired by imposing as little as 35% to 40% proximal stenosis. In further studies, these measurements appeared valid with either single or multiple stenoses, and the investigators proposed coronary flow reserve as a physiological measure that described the overall function of the artery as a unit (13,14).
coronary resistance vessel vasodilation can be compared while imposing various degrees of proximal epicardial conduit vessel stenosis. Using this technique and intra-coronary injections of radiographic contrast as a vasodilator stimulus, Gould et al. (13) demonstrated that coronary flow with unobstructed vessels increased four- to fivefold with maximal vasodilation. Although resting blood flow did not fall until an 85% proximal stenosis was imposed, the normal four- to fivefold coronary flow reserve was impaired by imposing as little as 35% to 40% proximal stenosis. In further studies, these measurements appeared valid with either single or multiple stenoses, and the investigators proposed coronary flow reserve as a physiological measure that described the overall function of the artery as a unit (13,14).
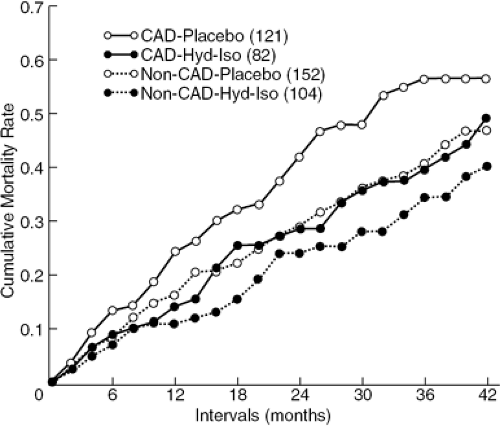 Figure 17-2 Cumulative mortality among patients with (n = 203) and without (n = 256) coronary artery disease (CAD) treated with placebo or hydralazine-isosorbide dinitrate (Hyd-Iso). (Reproduced from Tillisch J, Brunken R. Marshall R, et al. Reversibility of cardiac wall motion abnormalities predicted by positron tomography. N Engl J Med. 1986;314:884–888 .) |
In human studies, coronary flow reserve may be measured using subselective intracoronary Doppler flow velocity catheters. Although the Doppler flow catheter directly measures blood flow velocity, if the studies are performed with maximal vasodilation of the epicardial arteries, then changes in flow velocity are proportional to changes in flow volume. Sample Doppler tracings are shown in Figure 17-3. In addition, the combination of Doppler flow measurement guidewires and intravascular ultrasound (IVUS) catheters allows simultaneous direct measurement of coronary lumen diameter and flow velocity for calculation of coronary flow volumes. Using the Doppler technique alone, Wilson et al. demonstrated 3.5-fold increases over resting flow velocity with intracoronary radiographic contrast injections, fivefold increases with dipyridamole (15), and fivefold increases with papaverine (16) in patients with angiographically normal coronary arteries. These studies convincingly demonstrated coronary flow reserve in humans of a magnitude similar to that seen in the laboratory animal preparations described above.
Figure 17-4 illustrates the concept that as obstructive CAD progresses, coronary flow reserve declines to levels where ischemia and functional impairment occur with only modest increases in myocardial oxygen demand. In a variety of circumstances, the combined effect of atherosclerotic plaque and associated intravascular thrombus may reduce myocardial perfusion below levels required to prevent ischemia during daily activities or even at rest, and no coronary flow reserve exists.
Myocardial Dysfunction with Ischemia
When coronary flow reserve is inadequate, myocardial hypoperfusion results in cellular hypoxia. Because cardiac muscle is an obligate aerobic system with minimal anaerobic energy sources, this leads to biochemical, electrical, and mechanical dysfunction. This is summarized in Table 17-2.
Biochemical Dysfunction
Earlier studies of myocardial ischemia emphasized the rapid changes in intermediary metabolism that occur in association with ischemia. Jennings et al. studied high-energy phosphate metabolism after coronary occlusion in quick-frozen sections of canine hearts (17). They showed a shift from aerobic to anaerobic metabolism beginning within 8 to 10 seconds after coronary ligation. After 40 to 60 minutes of coronary ligation, their studies demonstrated marked loss of high-energy phosphate and low tissue pH, with characteristic ultrastructural changes, including cell swelling with mitochondrial and sarcolemma
damage. Clinical studies confirm that myocardial ischemia results in lactate production and a shift to free fatty acids (FFAs) as a preferred metabolic substrate.
damage. Clinical studies confirm that myocardial ischemia results in lactate production and a shift to free fatty acids (FFAs) as a preferred metabolic substrate.
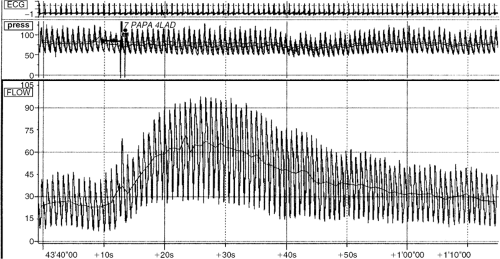 Figure 17-3 Coronary flow reserve (CFR) tracings in a patient with a nonobstructive left anterior descending (LAD) lesion. CFR = 66/21; LAD = 3.1. |
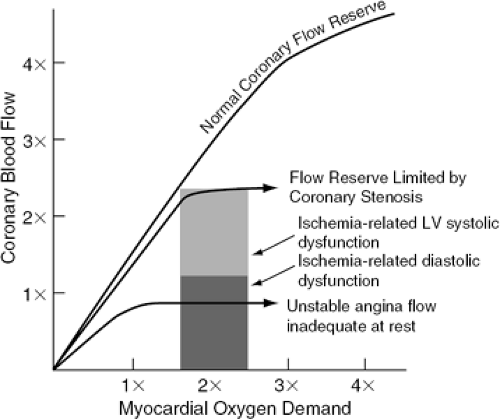 Figure 17-4 Decreasing coronary flow reserve with progressive coronary obstruction. |
Investigators using gene chip technology have characterized extensive changes in structural and regulatory proteins associated with myocardial ischemia and heart failure. The relatively large tissue samples removed at the time of left ventricular assist device (LVAD) implantation and acquisition of the explanted heart after transplant have facilitated these studies. Razeghi et al. from Houston have shown that mechanical unloading causes downregulation of numerous gene regulators of energy metabolism genes (18). Tan et al. at the Cleveland Clinic used gene chips to demonstrate unique patterns of gene expression in failing hearts (19). Margulies has recently reviewed a number of studies to support the concept that relief of mechanical loading on the heart triggers multiple signaling pathways that allow myocardial recovery (20).
Finally, large clinical trials have documented significant elevation of B-type natriuretic peptide (BNP) levels in acute coronary syndromes (21,22,23). The observed BNP levels can best be explained by increased synthesis and suggest that ischemia as well as mechanical loading stimulates BNP production (24).
In summary, profound metabolic changes occur with myocardial ischemia, including not only shifts in the substrates and products of energy metabolism, but also changes in structural and regulatory proteins and increases in BNP production.
Electrial Dysfunction
With the development of thrombolytic therapy and subsequently of primary angioplasty for management of acute coronary syndromes (ACS), reperfusion has displaced arrhythmia control as the focus of management. In the chronic setting, patients with left ventricular systolic dysfunction and ejection fraction (LVEF) less than 35% benefit from ICD devices, whereas patients with LVEF over 35% do not. These findings, and the association of larger fixed perfusion defects with inducible arrhythmia (25), suggest that scar, not ischemia, is the primary substrate for life-threatening arrhythmia in chronic CAD. Nonetheless, ischemia clearly increases susceptibility to arrhythmia. Stambler et al. (26) studied 20 patients with coronary disease who had no sustained ventricular tachycardia (VT) on baseline electrophysiological testing. With dobutamine infusion, induction of VT was associated with ischemia in nine subjects. The authors concluded that ischemia may contribute to ventricular arrythmia induction in some individuals. Ischemia increases QT dispersion, a marker for malignant ventricular arrhythmia (27), and preconditioning decreases QT dispersion (28). In an animal model, swine with total left anterior descending
coronary occlusions and hibernating myocardium had a high rate of sudden death due to ventricular fibrillation, suggesting that chronically ischemic myocardium results in “enhanced vulnerability to lethal arrhythmias and sudden cardiac death.” (29) In addition, Halkin et al. reported few cases of post-MI torsades de points that is related to an acquired long QT syndrome post-MI (30). Kenigsberg et al. reported that QT prolongation is the most consistent electro-cardiographic change seen in patients with balloon occlusion of the coronary artery (31). These data all suggest that, given a susceptible anatomic substrate, ischemia may indeed contribute to significant electrical instability.
coronary occlusions and hibernating myocardium had a high rate of sudden death due to ventricular fibrillation, suggesting that chronically ischemic myocardium results in “enhanced vulnerability to lethal arrhythmias and sudden cardiac death.” (29) In addition, Halkin et al. reported few cases of post-MI torsades de points that is related to an acquired long QT syndrome post-MI (30). Kenigsberg et al. reported that QT prolongation is the most consistent electro-cardiographic change seen in patients with balloon occlusion of the coronary artery (31). These data all suggest that, given a susceptible anatomic substrate, ischemia may indeed contribute to significant electrical instability.
Table 17-2 Myocardial Dysfunction with Ischemia | ||
|---|---|---|
|
Mechanical Dysfunction
The active energy requiring steps in myocyte contraction occur with transport of calcium back into the sarcoplasmic reticulum after actin—myosin interaction. Predictably, limited energy supplies first impair relaxation; only later does failure of contraction occur. In human studies, diastolic function appears to be more sensitive to ischemia than systolic function. Bonow et al. studied filling dynamics using radionuclide angiography in 26 patients with single-vessel CAD and normal left ventricular (LV) systolic function (32). After PTCA, the filling rate improved from 2.5 ± 0.6 to 3.0 ± 0.6 end-diastolic volume/sec and time to peak filling rate decreased from 178 ± 30 to 162 ± 20 milliseconds. These findings suggest that diastolic function may be measurably impaired while overall systolic function remains normal. Piscione et al. studied LV function during balloon occlusion of a coronary artery in 10 patients with normal LV function undergoing PTCA (33). With high-fidelity pressure records and LV angiography during the PTCA, they demonstrated impairment of relaxation within 20 seconds after onset of ischemia associated with impaired peak segmental shortening, as well.
In a dog model where ischemia was induced by 3 minutes of rapid pacing in the presence of moderate coronary stenosis, Takano and Glantz found that decreases in LV contractility were the best determinant of alterations in the LV end-diastolic pressure-volume relationship. They emphasized that coronary flow, myocardial supply and demand balance, and LV contractility (systolic function) were critical factors in altered diastolic function, while noting that in both canine and human hearts systolic function was only mildly depressed when the upward shift of the diastolic pressure-volume relationship was observed (34).
Ultimately, ischemia does depress systolic function. Development of a reversible LV wall motion disorder during exercise or pharmacologic stress as shown by echocardiography, radionuclide ventriculography, or magnetic resonance imaging (MRI) (35) represents the impairment of myocardial contraction associated with tissue hypoxia. Measures that include both the extent and severity of the wall motion abnormalities may improve the prognostic significance of stress echo studies (36).
Ischemic Preconditioning
Paradoxically, although ischemia produces significant impairment of cardiac function, repeated brief episodes of ischemia appear to protect against some of the detrimental consequences of ischemia-reperfusion injury (37,38,39). Among the mechanisms proposed to account for this phenomenon (ischemic preconditioning) are increases in endogenous adenosine, activation of potassium-adenosine triphosphate (K+-ATP) channels, and reductions in ischemia-related apoptosis. Preconditioning produced by episodes of tachycardia reduced infarct size in a canine model, and adenosine receptor blockade with 8-phenyltheophylline abolished the observed protective effects (40). Cleveland et al. confirmed the phenomenon of ischemic preconditioning in human right atrial tissue obtained at the time of surgery and demonstrated that long-term exposure to oral hypoglycemic agents prevented preconditioning (41). Their data suggest that preconditioning activates K+-ATP channels. Piot et al. have recently shown that ischemic preconditioning decreases apoptosis in rat hearts in vivo, lending additional support to the hypothesis that triggering of programmed cell death may be another important consequence of ischemia (42).
Nicoradil, a drug with both nitrate-like actions and K+-ATP channel-opener activity, appears to simulate the effects of preconditioning. Nicorandil is available in Europe and Japan, and many Japanese investigators have used the controlled circumstances of balloon angioplasty to evaluate the effects of nicorandil (43). Two recent trials, including 32 patients undergoing PTCA in one and 44 undergoing PTCA in the other, demonstrated enhanced myocardial tolerance to ischemia (typical of preconditioning) with nicorandil as compared to saline controls (44,45). The bulk of the nicorandil data support the concept that preconditioning involves activation of K+-ATP channels
Disturbingly, Dekker et al. (46) have reported a study of the effects of ischemic preconditioning on cellular electrical coupling in rabbit papillary muscle preparations from normal and failing hearts. In this model, ischemic preconditioning produced detrimental effects in failing papillary muscle, advancing the onset of irreversible damage. Aging (47) and diabetes (48) also appear to reduce the impact of preconditioning, although nicorandil may restore preconditioning responsiveness in the elderly (49). These observations suggest that the effects of repeated episodes of ischemic preconditioning may differ according to clinical circumstances.
In summary, under normal physiological circumstances myocardial blood flow exquisitely matches myocardial oxygen demand and can increase four- to fivefold in response to demand. As CAD diminishes physiological flow reserve, ischemia may result from either increases in oxygen demand or decreases in oxygen supply. Ischemia rapidly produces profound changes in intermediary energy metabolism, resulting in impaired diastolic and systolic function. In addition, chronic ischemia leads to altered gene expression, changes in structural and regulatory proteins, and initiation of cellular mechanisms leading to apoptosis and fibrosis. Furthermore, chronic ischemia causes increases in β-adrenergic receptor density, and both acute and chronic ischemia lead to increased myocardial production of BNP. In a self-protective adaptation, repeated brief episodes of ischemia may induce preconditioning, probably related to K+-ATP channel opening, reducing the extent of myocardial injury.
Myocardial Stunning and Hibernation
Myocardial stunning and hibernation, clinically important concepts in understanding heart failure due to CAD, represent a recent and dramatic paradigm shift in our approach to coronary disease. Before the introduction of these concepts, ischemic injury was thought of as an acute, self-limited process with only two possible outcomes: complete recovery of normal function or cellular necrosis with infarction and subsequent scar formation. In fact, ischemic LV dysfunction is far more complex. Impaired contraction may occur as a result of isolated or repetitive episodes of ischemia followed by reperfusion, as in unstable angina. This mechanical dysfunction is referred to as myocardial stunning. Contractile failure may also develop as a pathophysiological adaptation to chronic ischemia; the descriptive term hibernating myocardium has been used in this chronic setting (50,51,52,53,54,55).
As originally proposed, stunning implied a prolonged, albeit transient, state of impaired function following a momentary event. Imagine a boxer stunned after a violent blow to the head; within a few hours, he regains his full capacities. In contrast, hibernating indicated a prolonged state of metabolic downregulation with markedly reduced activity; for example, imagine a bear who will survive the long cold winter by sleeping curled in her den (56).
The concept of myocardial stunning is outlined in Figure 17-5. Myocardial stunning may occur after a single, prolonged episode or repetitive, brief episodes of coronary hypoperfusion, as demonstrated by increased diastolic stiffness after repeated balloon occlusions during PTCA. Wijns et al. (57) studied the effects of three to ten balloon inflations lasting 15 to 75 seconds on diastolic performance and demonstrated decreased diastolic function lasting up to 12 minutes after the final inflation, whereas systolic function returned quickly to normal. Impaired systolic function also occurs with repeated episodes of ischemia, as demonstrated by Nixon et al. (58). Stunned myocardium retains the ability to contract under inotropic stimulation or postextrasystolic potentiation (59). After successful thrombolysis, ejection fraction (EF) may remain depressed shortly after treatment but improves substantially 10 to 14 days later. Stack et al. (60) showed improvement in LVEF from 40% ± 8% to 48% ± 6% 16 days after successful lytic therapy; Reduto et al. (61) also demonstrated a similar improvement, 46% ± 14% to 55% ± 10% in 10 days.
Kim et al. have reviewed recent laboratory studies of the pathogenesis of stunning (62). In large-animal models, stunning produces significant changes in gene and protein regulation, with upregulation of an array of survival genes. Biopsy specimens from patients undergoing cardiac surgery also suggest important differences in stunned and hibernating tissue as compared to normal control samples, with increased cyclic adenosine monophosphate (cAMP) in hibernating myocardium and increased levels of heat shock protein 72 in stunned tissue (63). These data demonstrate that, at a very basic level, stunning produces more profound changes than just a brief alteration in intermediary metabolism; in fact, gene expression is altered to favor protection of the threatened myocardium.
Hibernating myocardium describes chronic contractile dysfunction at rest occurring in response to marked impairment of flow reserve. Resting myocardial blood flow in hibernating segments is close to normal, but extremely limited flow reserve leads to myocardial ischemia with minimal activity and to chronic ventricular dysfunction (64). The dysfunction is reversible by revascularization (65). This concept is diagrammed in Figure 17-6. Keller et al. have studied the effects of reduced coronary flow on myocardial metabolism in the isolated perfused rat heart using nuclear magnetic resonance spectroscopy (66). Their data indicate that modest decreases in coronary flow reduce contractile performance and myocardial oxygen consumption before any observable decrease in adenosine triphosphate (ATP) or myocardial pH occurs, and before significant lactate production begins. These balanced reductions in ventricular performance and oxygen consumption without the traditional metabolic markers of ischemia were felt to provide a model for hibernating myocardium, in that reduction of contractility appeared to be directly related to reduced tissue oxygen delivery.
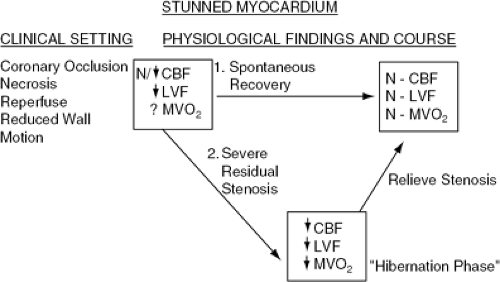 Figure 17-5 When a severe coronary stenosis reduces coronary flow without necrosis, there may be a downregulation of myocardial function. The decrease in myocardial contractility reduces myocardial energy demands; hence, no necrosis occurs. Thus, the physiologic findings in the course of this clinical phenomenon would be reduced coronary blood flow, reduced LV function, and reduced myocardial oxygen demands. There are three possible consequences given these findings. The first, spontaneous recovery with which coronary blood flow may normalize due to collateral flow relief of coronary spasm, will result in return of LV myocardial oxygen requirements and function. The second possibility is to relieve the stenosis. Under these circumstances coronary blood flow normalizes immediately, but LV function may recover immediately or its recovery may be delayed. If its recovery is delayed, the findings (basically, a normal coronary blood flow with reduced LV function and, presumably, myocardial oxygen demand) resemble those seen in myocardial stunning, which occurs after MI is reprefused. Thus, the hibernating myocardium may go through a delayed recovery phase, which we have termed a stunned phase. With time, spontaneous recovery should occur. A third possibility would be if the relationship between the reduced coronary blood flow and reduced myocardial oxygen demand is disturbed by further reduction in coronary blood flow for an increase in myocardial oxygen consumption; myocardial necrosis may ensue. Under these circumstances the necrosed myocardium may be reperfused, in which case coronary blood flow is normalized; this myocardial region also may go through a stunned phase before spontaneous recovery. N, normal. |
Evidence confirming that restoration of flow to hibernating myocardium results in restoration of function continues to accumulate (67). Topol et al. demonstrated improved wall thickening by transesophageal echo immediately after surgical revascularization in 20 patients undergoing CABG
(68). Tillisch et al. showed improved wall motion after revascularization in 35 of 41 ischemic myocardial segments with preserved glucose uptake on positron emission tomography, whereas 24 of 26 segments without glucose uptake failed to improve (69). In a 3-year follow-up study of 70 patients with CAD and depressed LV function who underwent CABG, Pagley et al. found that the extent of viable myocardium, as determined by preoperative thallium 201 scintigraphy, was the only independent predictor of cardiac event-free survival (70). Meluzin et al. showed that in revascularized patients with CAD and moderate or severe LV dysfunction, the presence of a large amount of dysfunctional but viable myocardium identified patients with the best prognosis (71) (Fig. 17-7). In a meta-analysis of 24 studies involving 3,088 patients with depressed LV function, patients with documented viability by thallium perfusion imaging, positron emission tomography (PET) scanning, or dobutamine echocardiography had a significant 80% reduction in annual mortality with revascularization (3.2% versus 16% for medical therapy). The degree of LV dysfunction was directly related to the magnitude of benefit (72).
(68). Tillisch et al. showed improved wall motion after revascularization in 35 of 41 ischemic myocardial segments with preserved glucose uptake on positron emission tomography, whereas 24 of 26 segments without glucose uptake failed to improve (69). In a 3-year follow-up study of 70 patients with CAD and depressed LV function who underwent CABG, Pagley et al. found that the extent of viable myocardium, as determined by preoperative thallium 201 scintigraphy, was the only independent predictor of cardiac event-free survival (70). Meluzin et al. showed that in revascularized patients with CAD and moderate or severe LV dysfunction, the presence of a large amount of dysfunctional but viable myocardium identified patients with the best prognosis (71) (Fig. 17-7). In a meta-analysis of 24 studies involving 3,088 patients with depressed LV function, patients with documented viability by thallium perfusion imaging, positron emission tomography (PET) scanning, or dobutamine echocardiography had a significant 80% reduction in annual mortality with revascularization (3.2% versus 16% for medical therapy). The degree of LV dysfunction was directly related to the magnitude of benefit (72).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


