1
7.4.6 GPCR Kinase
7.4.7 Cardiorenal Syndrome
7.5.1 Modeling
7.6 Remodeling
7.6.3 Cardiac Hypertrophy
7.6.4 Cardiac Fibrosis
7.6.6 Sarcomeric Constituents
7.7 Anemia
7.8 Cardiac Cachexia
7.9.2 Calcium
7.9.3 Renin–Angiotensin Axis
7.9.4 Natriuretic Peptides
7.9.5 Endothelin-1
7.9.6 Relaxins
7.9.7 Vascular Endothelium
7.9.10 Periostin
7.9.11 Nucleotides
7.9.12 Small GTPases
7.9.13 Kinases and Phosphatases
7.9.15 Inflammatory Cytokines
7.9.16 Chemokines
7.9.17 MicroRNAs in Cardiac Failure
According to the New Encyclopædia Britannica (15th ed., 1985), “heart failure is the inability of either or both side of the heart to pump sufficient blood to meet the needs of the body…. Heart failure is characterized by distension of the veins serving the lungs or of those serving the rest of the body, or of both; this engorgement is expressed in the term congestive heart failure.”
The inappropriate pumping results from myocardial weakness. The structure–function relation of the heart is impaired and bodily needs are unmatched. Cardiac dysfunction is defined as an alteration in the relation between preload and stroke volume. Heart failure is also frequently associated with arrhythmias and valve dysfunction.
Heart failure develops progressively after cardiac damages. It can affect the right or left pump only or, most often, the entire heart. Clinical manifestations comprise fatigue, dyspnea (breathlessness), diminished exercise capacity, and fluid retention.
Heart failure causes significant morbidity and mortality. It currently affects 0.4–2 % of the population in the Western world and ranges from 6 to 10 % patients over 65 years old, with a 4-year survival rate of 50 %. Heart failure is a frequent cause of atrial fibrillation and patients often die from sudden arrhythmia.
Diagnosis relies on either two major criteria, such as elevated jugular venous pressure, pulmonary rates, or a third heart sound, or one major criterion and two minor criteria, such as edema, dyspnea, or hepatomegaly. Acute episodes cause cardiac decompensation.
Echocardiography and magnetic resonance and nuclear medical imaging are commonly employed in the identification of ventricular systolic dysfunction.
Heart failure is not a simple contractile disorder or a single disease of the heart. It can result from maladaptive cardiac remodeling in response to pressure or volume overloading in the acute phase of cardiac injury, such as in mitral insufficiency and myocardial infarction, that leads to progressive decompensation.
Adverse cardiac remodeling is defined as an alteration in the shape, dimensions, structure, and mass of the heart in response to hemodynamic load and/or cardiac injury. Adverse cardiac remodeling evolves in a neurohormonal activation context (Chap. 3).
Whatever the cause, heart failure is associated with a chronic activation of β-adrenoceptors and elevated plasma catecholamine levels (Sect. 7.9.1). Initially, a hyperactive sympathetic nervous system is aimed at compensating contractile dysfunction. However, β-adrenoceptor overstimulation provokes cardiac hypertrophy and arrhythmias, hence progressively disorganizing the cardiac structure and deteriorating its contractile function.
Therapy of heart failure is aimed at correcting the reduced blood flow. Antineuroendocrine treatment with angiotensin-converting enzyme inhibitors, β-adrenergic blocking agents, and antialdosterone drugs can significantly reduce morbidity and mortality [208].
Therapy also targets adverse cardiac remodeling. Reverse remodeling can result from pharmacological and nonpharmacological therapies. Moderate endurance training and continuous positive airway pressure (CPAP) therapy in heart failure patients with sleep apnea induce reverse remodeling [209]. Cardiac resynchronization improves exercise capacity and quality of life in patients with ventricular dyssynchrony. Reverse remodeling by surgery, such as mitral valve replacement, aneurysmectomy, and volume reduction are associated with high perioperative mortality. Mechanical unloading of the failing ventricles by left ventricular assist systems reverses remodeling on the cellular and subcellular levels. New therapeutic procedures, such as gene or stem cell therapy, in reverse remodeling await characterization.
Heart failure with preserved left ventricular ejection fraction (HFpLVEF; with normal or quasinormal systolic function) is nearly as deadly as heart failure with reduced left ventricular ejection fraction (HFrLVEF). Metabolic risk factors of HFpLVEF include diabetes, obesity (hyperlipidemia), and hypertension.
Inhibition of the sympathetic nervous system and renin–angiotensin–aldosterone axis is effective in the treatment of HFrLVEF patients. However, medication used in HFrLVEF patients (e.g., β-blockers and angiotensin-converting enzyme inhibitors) are not efficient in HFpLVEF patients [222].
In fact, patients can be classified into three groups based on review of the left ventricular ejection fraction in the medical record [508]: (1) patients with persistently preserved (EF ≥40 %; HFpEF); (2) with recovered (EF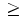 40 %; HFrEF); and (3) with low ejection fraction (EF
40 %; HFrEF); and (3) with low ejection fraction (EF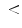 40 %; HFlEF). HFrEF patients have less atrial fibrillation episodes, hypertension, and diabetes, better renal function, and larger end-diastolic left ventricular size than HFpEF patients. They are younger than HFpEF and HFlEF patients. They have lower rates of coronary artery disease than HFlEF patients.
40 %; HFlEF). HFrEF patients have less atrial fibrillation episodes, hypertension, and diabetes, better renal function, and larger end-diastolic left ventricular size than HFpEF patients. They are younger than HFpEF and HFlEF patients. They have lower rates of coronary artery disease than HFlEF patients.
40 %; HFrEF); and (3) with low ejection fraction (EF 40 %; HFlEF). HFrEF patients have less atrial fibrillation episodes, hypertension, and diabetes, better renal function, and larger end-diastolic left ventricular size than HFpEF patients. They are younger than HFpEF and HFlEF patients. They have lower rates of coronary artery disease than HFlEF patients.7.1 Risk Factors, Major Causes, and Natural History of Cardiac Deficiency
The likelihood of developing heart failure rises in the presence of:
Hypertension;
Obstructive coronary artery disease;
Cardiomyopathies of nonischemic origin, such as idiopathic dilated cardiomyopathy;
Cardiac rhythm and conduction disorders;
Endo- or myocarditis; and
Cardiac valve diseases.
Three main types of heart failure exist:
Heart failure due to left ventricular weakness;
Heart failure with preserved ejection fraction usually caused by a stiff left ventricle; and
Heart failure due to valve disease.
Many etiologies can lead to the onset of heart failure. Cardiac causes of acute decompensated heart failure, or acute dyspnea, include left ventricular diastolic and/or systolic dysfunction, right ventricular dysfunction, and/or acute ischemia.
Chronic heart failure results from:
1.
Cardiac disorders, such as acute and chronic ischemic heart disease, valvular disorders, cardiomyopathies, myocarditis, and chronic arrhythmias, among others;
2.
External factors, such as hypertension, pulmonary embolism, and shunt, among others; and
3.
Defects in genes that encode constituents of the sarcomere, cytoskeleton, mitochondrion, or extracellular matrix.
According to the American Heart Association and American College of Cardiology several stages of latent or patent heart failure can be defined [480]:
Stage A
representing patients at risk of heart failure, without structural heart disease or symptoms of heart failure;
Stage B
characterized by patients with structural heart disease, but without symptoms of heart failure
Stage C
defined by patients with structural heart disease and past or present symptoms of heart failure; and
Stage D
corresponding to patients with advanced (refractory) heart failure requiring specialized interventions (e.g., inotropic support).
7.2 Structure and Function of Chronic Congestive Heart Failure
Congestive heart failure, the final stage of many diseases of the heart, is a weakening of the myocardium that fails to supply adequate blood flow to the bodily organs. The ineffective blood pumping is associated with activated stress pathways by mechanical and chemical signals, myocardial dysfunction, cell death, and altered cell environment.
Chronic heart failure is linked to myocardial structural and functional abnormalities. The mass of failing hearts increases due to adverse cardiomyocyte hypertrophy as well as remodeling with fibrosis, necrosis, and cardiac dilation. Edema of the myocardial interstitium increases wall stiffness and causes collagen deposition leading to fibrosis.
Heart failures related to a preserved or reduced left ventricular ejection fraction have similar outcomes [509].
Diastolic dysfunction results from myocardial remodeling, that is, cardiomyocyte hypertrophy and interstitial fibrosis. It is linked to altered relaxation (prolonged isovolumic relaxation and deceleration times and abnormal left ventricular filling) and cardiac wall stiffness (or reduced left ventricular compliance), with elevated diastolic pressure, enlarged left atrial volume and reduced emptying (A wave), whereas systolic function is preserved. It leads to heart failure with preserved left ventricular ejection fraction. Diastolic dysfunction increases the risk of mortality. Hypophosphorylation of titin that intervenes in serves in distensibility and diastolic recoil of cardiomyocytes can lead to HFpLVEF associated with hypertrophy in patients [223].
Systolic heart failure is characterized by contractile failure and dilated heart chamber;
Diastolic heart failure by a preserved ejection fraction, heart contractility being considered as normal, and wall thickening (cardiac hypertrophy).
Chronic heart failure triggers general and regional neurohormonal compensatory responses, such as activation of the sympathetic nervous system, renin–angiotensin–aldosterone axis, nonosmotic release of vasopressin, and liberation of natriuretic peptides, endothelial regulators such as nitric oxide, and cytokines. These countermeasures include retention of sodium and water by the kidney, release of neurohormones, and activation of various intracellular signaling cascades in cells of the cardiovascular system.
The long-term activation of these compensatory mechanisms initially aimed at preserving hemodynamics lead progressively to cardiac dilation and decaying contractility down to decompensated stage.
Chronic heart failure is associated with impaired myocardial metabolism and signaling, altered cardiac receptors and contractile proteins, defective calcium handling, sodium retention, neurohumoral maladaptations, oxidative stress, and vascular dysfunction.
Cardiomyocytes reply to stress and abnormal loading by reprogramming gene expression, especially genes involved in metabolism and ion handling as well as genes specific of embryo- and fetogenesis.
7.3 Markers of Chronic Heart Failure
Numerous markers related to heart failure explore the role of inflammation, oxidative stress, cardiac remodeling, cardiomyocyte stretching, and renal function, among others. The most appropriate markers are selected for diagnosis, prognosis, and treatment planning. Concentrations of circulating troponins increase not only in myocardial infarction upon atherosclerotic plaque ulceration, fissuring, and rupture (type-1 myocardial infarction), as well as supply–demand imbalance and endothelial dysfunction (type-2 myocardial infarction), but also in acute and chronic heart failure, in particular due to cardiomyocyte apoptosis and autophagy subsequently to wall stretch, arrhythmia, infiltration such as amyloidosis, exposure to toxics (e.g., alcohol or chemotherapy drugs), myocarditis, and stress cardiomyopathy, as well as renal dysfunction [510]. Elevated concentrations of troponin-I or -T predicts an adverse outcome. Natriuretic peptides are currently reference markers for diagnosis and therapy monitoring of heart failure. However, natriuretic peptides are secreted in response to a nonspecific pressure overload of any cardiac chamber that can be observed not only in heart failure but also in systemic hypertension, atrial fibrillation, and valvular disease, as well as lung diseases, pulmonary embolism, and renal failure [511]. Therefore, natriuretic peptide marker specificity for diagnosis is low. However, a BNP level in the normal range excludes heart failure as the cause of complaints such as dyspnea.
In addition, the BNP release results from a neurohormomal activation cascade aimed at compensating a anatomical and/or functional defect and yields a prognosis information. Although BNP exerts vasodilatory and diuretic effects, an increase in BNP level is associated with a worse outcome in heart failure [511]. Therefore, the measurement of natriuretic peptides (type-B natriuretic peptide [BNP] and N-terminally truncated proBNP [proBNP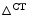 ]), a marker for cardiomyocyte stretching, as well as mid-regional proatrial natriuretic peptide [proANP
]), a marker for cardiomyocyte stretching, as well as mid-regional proatrial natriuretic peptide [proANP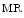 ]) is recommended to exclude alternative causes of dyspnea in patients with suspected heart failure and to evaluate the prognosis.
]) is recommended to exclude alternative causes of dyspnea in patients with suspected heart failure and to evaluate the prognosis.
]), a marker for cardiomyocyte stretching, as well as mid-regional proatrial natriuretic peptide [proANP]) is recommended to exclude alternative causes of dyspnea in patients with suspected heart failure and to evaluate the prognosis.Quiescin Q6 sulfhydryl oxidases (QSOx) are thiol oxidases involved in oxidative protein folding with thiol–disulfide exchange, cell cycle control, and extracellular matrix remodeling.1 They catalyzes the insertion of disulfide bonds into unfolded reduced proteins with concomitant reduction of oxygen to hydrogen peroxide, hence altering the redox environment:2
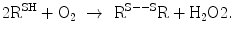
Quiescin Q6 sulfhydryl oxidase QSOx1 identifies acute decompensated heart failure (ADHF), particularly when combined with BNP and proBNP [513].
[513].
(7.1)
[513].The QSOx1 synthesis rate in the left ventricle and left atrium depends on the degree of pressure overload. Myocardial QSOx1 expression remains low in patients with acute dyspnea of noncardiac origin or with stable compensated heart failure.
In addition to C-reactive protein, pentraxin-3, and interleukin-6, tumor-necrosis factor-α receptor TNFRSF1a and growth differentiation factor GDF5 are markers of inflammation. Marker levels are linked to the pathophysiological mechanisms of heart failure, e.g. heart failure with reduced or preserved left ventricular ejection fraction [511]. With increasing age, the level of expression of inflammatory markers rises in the general population.
Markers related to extracellular matrix remodeling comprise syndecan-1 and periostin [514].
Disparities in pathophysiology and disease manifestation exist between women and men with heart failure. TNFRSF1a, GDF15, and proBNP show the strongest interaction between sex and mortality, mortality being lower in women than men, independently of differences in clinical characteristics [514]. The main etiological sex difference in heart failure is a predominance of myocardial infarctions and the presence of ischemic heart disease in men with respect to women. However, most comorbidities are preferentially observed in women with heart failure. In addition, in men, remodeling is usually characterized by left ventricular dilation and fibrosis, whereas in women, the heart more frequently remodels with a marked concentric hypertrophy and a smaller left ventricular volume [514].
show the strongest interaction between sex and mortality, mortality being lower in women than men, independently of differences in clinical characteristics [514]. The main etiological sex difference in heart failure is a predominance of myocardial infarctions and the presence of ischemic heart disease in men with respect to women. However, most comorbidities are preferentially observed in women with heart failure. In addition, in men, remodeling is usually characterized by left ventricular dilation and fibrosis, whereas in women, the heart more frequently remodels with a marked concentric hypertrophy and a smaller left ventricular volume [514].
show the strongest interaction between sex and mortality, mortality being lower in women than men, independently of differences in clinical characteristics [514]. The main etiological sex difference in heart failure is a predominance of myocardial infarctions and the presence of ischemic heart disease in men with respect to women. However, most comorbidities are preferentially observed in women with heart failure. In addition, in men, remodeling is usually characterized by left ventricular dilation and fibrosis, whereas in women, the heart more frequently remodels with a marked concentric hypertrophy and a smaller left ventricular volume [514].7.4 Genetic Background and Chronic Heart Failure
Heart failure has a strong heritable component. Parental heart failure confers a 70 % greater disease risk than the absence of such a family history. Traditionally, inherited genetic forms of heart failure are distinguished from acquired forms. The former category encompasses familial dilated, hypertrophic, and arrhythmogenic right ventricular cardiomyopathy (Chap. 4).
Mutations of numerous genes that cause inherited forms of heart failure encode proteins that function in contractility (sarcomeric, cytoskeletal, and desmosomal proteins, as well as lamin-A and -C), energy production (mitochondrial proteins), calcium handling, and transcription regulation (Table 7.1). In addition, reprogramming of gene expression represents the response of cardiomyocytes exposed to stress (heart failure dysregulome).
Table 7.1
Inherited forms of heart failure (Source: [515]). Differences in contractile and regulatory function exist among isoforms of myosin, tropomyosin, and troponin-T. Mutations in a given gene causes different forms of genetic cardiomyopathies
Gene protein | Expression in heart failure |
|---|---|
Sarcomeric constituents | |
ACTC1 | ↑ |
MYH6 | ↓ |
MYH7 | ↑ |
TNNC1, TNNI3, TNNT2 | ↑ |
TPM1/2 | ↑ |
MYBPC3 | Similar |
Actinin-α | ↑ |
Titin | Similar |
Costameric constituents | |
Desmin | ↑ |
Dystrophin | ND |
Metavinculin | ND |
Muscle LIM protein | ↓ |
Sarcoglycan-δ | Similar |
Nuclear constituents | |
Lamin-A/C | ↓ |
Energetic metabolism components | |
Succinate deshydrogenase | ND |
Ion handling components | |
Phopholamban | ↑ |
ABCC9 | ND |
SCN5A | ↓ |
Miscellaneous | |
ANKRD1 | ↑ |
EYA4 | ND |
Ldb3 | ND |
Rbm20 | ND |
TAZ | ND |
TMPO | ND |
The contraction and relaxation at relatively high frequency (∼ 1 Hz) of cardiomycytes are mediated by highly coordinated processes that allow rapid activation and deactivation of the sarcomeric filaments. The sarcomere is the fundamental contractile unit. Its shortening results from arrangements of myosin microfilaments sliding on arrays of actin microfilaments fueled by ATP hydrolysis with a cyclic ratchet-like interaction regulated predominantly by the actin filament regulatory partners troponin and tropomyosin.
A change in myofilament isoform composition affects myocardial contractile performance in heart failure. The fast isoform of myosin heavy chain α MHC (MyHC6) accounts for approximately 10 % of the content in the healthy human cardiomyocyte myocardium (hence 90 % β MHC or MyHC7) [516]. This proportion is further reduced to nearly undetectable concentrations during heart failure, thereby lowering cardiac function. Furthermore, a shift in troponin-T isoform composition as well as in tropomyosin isoform expression also occurs in the human failing heart.
7.4.1 Troponins
Among striated myocytes, the regulation of cardiomyocyte contraction differ from that of skeletal myocytes due to distinct physiological and contractile properties. The troponin complex is a component of thin actin filaments with tropomyosin. Troponin contains three subunits:
Troponin binds to tropomyosin and, in the absence of calcium, also to actin via troponin-I.
1.
The calcium-binding troponin-C,
2.
The inhibitory troponin-I, and
3.
The tropomyosin-binding troponin-T.
The actin–myosin interaction is activated by calcium binding to troponin-C that provokes a conformational change in troponin, thereby decreasing troponin-I affinity to actin and engendering an azimuthal shift of tropomyosin on the actin filament that partly exposes myosin-binding sites on actin. Binding of one myosin to actin filament shifts tropomyosin further on actin and fully exposes the adjacent actin-binding sites and accelerates the rate of subsequent myosin binding to actin filament. In addition, myosin binding to actin increases the calcium affinity of unsaturated troponin-C, thereby potentiating actin activation [516].
Multiple cardiac troponin-T (cTnnT) isoforms are expressed in a developmentally regulated manner, whereas cardiac troponins cTnnC and cTnnI are expressed as single isoforms in the adult heart.
Contraction of the cardiomyocyte is carried out in many steps (Vol. 5, Chap. 5. Cardiomyocytes):
The rate of transition from weak to strong cross-bridge is not only influenced by myosin features but also by the Ca -binding properties of troponin-C on the thin actin filament as well as the level of free Ca
-binding properties of troponin-C on the thin actin filament as well as the level of free Ca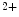 ions [517]. The rate of contraction can rise up to fivefold with increasing Ca
ions [517]. The rate of contraction can rise up to fivefold with increasing Ca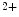 concentration up to a saturating amount. The rate of contraction at submaximal concentrations of Ca
concentration up to a saturating amount. The rate of contraction at submaximal concentrations of Ca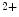 is modulated by the Ca
is modulated by the Ca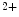 sensitivity of troponin-C. The myosin isozymes and ATP hydrolysis products modulate the rate of cardiomyocyte contraction independently of troponin-C. The intrinsic properties of myosin dictate the rate of contraction, with α-myosin having a faster ATPase cycle than that of β-myosin. The products of ATP hydrolysis inorganic phosphate (P
sensitivity of troponin-C. The myosin isozymes and ATP hydrolysis products modulate the rate of cardiomyocyte contraction independently of troponin-C. The intrinsic properties of myosin dictate the rate of contraction, with α-myosin having a faster ATPase cycle than that of β-myosin. The products of ATP hydrolysis inorganic phosphate (P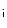 ) and ADP increase and decrease the maximal rate of myosin ATPase activity, respectively.
) and ADP increase and decrease the maximal rate of myosin ATPase activity, respectively.
1.
Increase in cytosolic Ca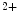 concentration;
concentration;
concentration;2.
Ca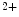 binding to troponin-C;
binding to troponin-C;
binding to troponin-C;3.
Troponin-C tethering to troponin-I;
4.
Troponin-I dissociation from actin;
5.
Tropomyosin position on actin shift;
6.
Weakly bound cross-bridges transition to strongly bound cross-bridges further moving tropomyosin on actin (a rate-limiting step for contraction); and
7.
Sarcomere shortening.
-binding properties of troponin-C on the thin actin filament as well as the level of free Ca ions [517]. The rate of contraction can rise up to fivefold with increasing Ca concentration up to a saturating amount. The rate of contraction at submaximal concentrations of Ca is modulated by the Ca sensitivity of troponin-C. The myosin isozymes and ATP hydrolysis products modulate the rate of cardiomyocyte contraction independently of troponin-C. The intrinsic properties of myosin dictate the rate of contraction, with α-myosin having a faster ATPase cycle than that of β-myosin. The products of ATP hydrolysis inorganic phosphate (P) and ADP increase and decrease the maximal rate of myosin ATPase activity, respectively.Relaxation of the cardiomyocyte involves several rate-limiting steps [517]:
Cytosolic metabolites that affect the cross-bridge detachment kinetics modify the rate of relaxation independently of the fall in cytosolic Ca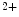 level. Increase in ADP concentration diminishes cross-bridge detachment and the rate of relaxation, whereas increase in P
level. Increase in ADP concentration diminishes cross-bridge detachment and the rate of relaxation, whereas increase in P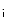 concentration heightens cross-bridge detachment and the rate of relaxation. Change in myosin heavy chain expression also modulates the rate of cardiomyocyte relaxation. In addition, β-adrenoceptor stimulation accelerates the rate of relaxation, as it raises the rate of Ca
concentration heightens cross-bridge detachment and the rate of relaxation. Change in myosin heavy chain expression also modulates the rate of cardiomyocyte relaxation. In addition, β-adrenoceptor stimulation accelerates the rate of relaxation, as it raises the rate of Ca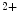 dissociation from troponin-C via phosphorylation of cTnnI by PKA. On the other hand, in TNNC1 gene mutation related to familial hypertrophic cardiomyopathy, increased Ca
dissociation from troponin-C via phosphorylation of cTnnI by PKA. On the other hand, in TNNC1 gene mutation related to familial hypertrophic cardiomyopathy, increased Ca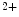 sensitivity attenuates the rate of relaxation.
sensitivity attenuates the rate of relaxation.
1.
Decline in cytosolic Ca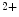 concentration;
concentration;
concentration;2.
Transient dissociation of Ca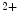 from troponin-C; and
from troponin-C; and
from troponin-C; and3.
Cross-bridge detachment.
level. Increase in ADP concentration diminishes cross-bridge detachment and the rate of relaxation, whereas increase in P concentration heightens cross-bridge detachment and the rate of relaxation. Change in myosin heavy chain expression also modulates the rate of cardiomyocyte relaxation. In addition, β-adrenoceptor stimulation accelerates the rate of relaxation, as it raises the rate of Ca dissociation from troponin-C via phosphorylation of cTnnI by PKA. On the other hand, in TNNC1 gene mutation related to familial hypertrophic cardiomyopathy, increased Ca sensitivity attenuates the rate of relaxation.Slow and fast calcium-binding troponin-C are encoded by the TNNC1 and TNNC2 genes, respectively. The TnnC1 isoform corresponds to the cardiac (cTnnC) subtype. It possesses an N and C lobe. The former is the regulatory part that binds to Ca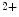 ion and troponin-I after calcium binding, hence regulating calcium-regulated myofibrillar ATPase activity. The latter binds Ca
ion and troponin-I after calcium binding, hence regulating calcium-regulated myofibrillar ATPase activity. The latter binds Ca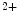 or Mg
or Mg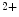 ions.
ions.
ion and troponin-I after calcium binding, hence regulating calcium-regulated myofibrillar ATPase activity. The latter binds Ca or Mg ions.Cardiac troponin-C (cTnnC) is the major regulator of myocardial contraction. The TnnC1 subtype, a member of the EF-hand family of Ca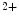 -binding proteins, relays the Ca
-binding proteins, relays the Ca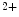 signal upon acute brief Ca
signal upon acute brief Ca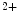 influx (i.e., Ca
influx (i.e., Ca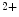 entry and release from the extracellular space and intracellular stores, respectively) via a conformational change to the rest of the troponin–tropomyosin complex. It possesses different functional and Ca
entry and release from the extracellular space and intracellular stores, respectively) via a conformational change to the rest of the troponin–tropomyosin complex. It possesses different functional and Ca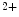 -binding properties compared with skeletal subtypes (skTnnC). Upon Ca
-binding properties compared with skeletal subtypes (skTnnC). Upon Ca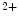 binding, the regulatory N-domain of skTnnC switches from a closed to an open conformation, thereby exposing a patch of hydrophobic residues and hence interacting with skTnnI. Unlike, other EF-hand Ca
binding, the regulatory N-domain of skTnnC switches from a closed to an open conformation, thereby exposing a patch of hydrophobic residues and hence interacting with skTnnI. Unlike, other EF-hand Ca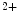 -binding calcium sensors skTnnC and calmodulin characterized by a large conformational change upon Ca
-binding calcium sensors skTnnC and calmodulin characterized by a large conformational change upon Ca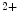 binding as well as the EF-hand calcium buffers parvalbumin and calbindin characterized by a minor conformational changes upon Ca
binding as well as the EF-hand calcium buffers parvalbumin and calbindin characterized by a minor conformational changes upon Ca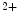 tethering, cTnnC has a compact regulatory domain in the Ca
tethering, cTnnC has a compact regulatory domain in the Ca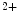 -bound structure [518]. The free energy of Ca
-bound structure [518]. The free energy of Ca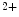 binding to the cTnnC-cTnnI complex is four times smaller than that to the skTnnC-skTnnI complex that enables the repetition at relatively high frequency of contraction–relaxation cycles.
binding to the cTnnC-cTnnI complex is four times smaller than that to the skTnnC-skTnnI complex that enables the repetition at relatively high frequency of contraction–relaxation cycles.
-binding proteins, relays the Ca signal upon acute brief Ca influx (i.e., Ca entry and release from the extracellular space and intracellular stores, respectively) via a conformational change to the rest of the troponin–tropomyosin complex. It possesses different functional and Ca-binding properties compared with skeletal subtypes (skTnnC). Upon Ca binding, the regulatory N-domain of skTnnC switches from a closed to an open conformation, thereby exposing a patch of hydrophobic residues and hence interacting with skTnnI. Unlike, other EF-hand Ca-binding calcium sensors skTnnC and calmodulin characterized by a large conformational change upon Ca binding as well as the EF-hand calcium buffers parvalbumin and calbindin characterized by a minor conformational changes upon Ca tethering, cTnnC has a compact regulatory domain in the Ca-bound structure [518]. The free energy of Ca binding to the cTnnC-cTnnI complex is four times smaller than that to the skTnnC-skTnnI complex that enables the repetition at relatively high frequency of contraction–relaxation cycles.Calcium binding to and dissociation from cTnnC modulate the cardiomyocyte contraction and relaxation. Calcium removal rate of cTnnC is about threefold faster than that of skTnnC. The subsequent cTnnC–cTnnI interaction determines the specific regulation of cardiomyocyte contraction. However, actin–tropomyosin and myosin influence Ca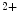 unbinding from troponin, actin–tropomyosin increasing the rate of Ca
unbinding from troponin, actin–tropomyosin increasing the rate of Ca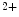 dissociation from 42 to 105/s and myosin slowing it to 13/s [517]. Mutations in troponin subunit genes linked to cardiomyopathies can sensitize or desensitize cTnnC to Ca
dissociation from 42 to 105/s and myosin slowing it to 13/s [517]. Mutations in troponin subunit genes linked to cardiomyopathies can sensitize or desensitize cTnnC to Ca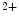 and/or affect the rates of Ca
and/or affect the rates of Ca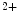 exchange with cTnnC. Slow skeletal TnnI (sskTnnI) expressed in embryonic and early postnatal hearts and possibly reexpressed during disease increases the Ca
exchange with cTnnC. Slow skeletal TnnI (sskTnnI) expressed in embryonic and early postnatal hearts and possibly reexpressed during disease increases the Ca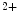 affinity and decreases the rate of Ca
affinity and decreases the rate of Ca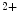 dissociation from cTnnC in the troponin complex on the actin filament in the presence of myosin [517].
dissociation from cTnnC in the troponin complex on the actin filament in the presence of myosin [517].
unbinding from troponin, actin–tropomyosin increasing the rate of Ca dissociation from 42 to 105/s and myosin slowing it to 13/s [517]. Mutations in troponin subunit genes linked to cardiomyopathies can sensitize or desensitize cTnnC to Ca and/or affect the rates of Ca exchange with cTnnC. Slow skeletal TnnI (sskTnnI) expressed in embryonic and early postnatal hearts and possibly reexpressed during disease increases the Ca affinity and decreases the rate of Ca dissociation from cTnnC in the troponin complex on the actin filament in the presence of myosin [517].Distinct genes encode cardiac-specific (TNNI3) and fast (TNNI2) and slow (TNNI1) skeletal muscle-specific isoforms of inhibitory troponin-I. Mutations of the TNNI3 gene linked to hypertrophic or restrictive cardiomyopathies (R145G, R145W, D190H, and R192H) increase the Ca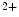 sensitivity of the troponin complex on the actin filament (but not in isolated cTnnI) [517].
sensitivity of the troponin complex on the actin filament (but not in isolated cTnnI) [517].
sensitivity of the troponin complex on the actin filament (but not in isolated cTnnI) [517].Tissue-specific subtypes of troponin-T that forms the troponin–tropomyosin complex include slow (TnnT1) and fast (TnnT3) skeletal muscle troponin-T encoded by the TNNT1 and TNNT3 genes, respectively, and cardiac troponin-T2 encoded by the TNNT2 gene.
In the human heart, four cardiac troponin-C isoforms with different calcium sensitivity are produced (cTnnT1–cTnnT4) from alternative splicing (cTnnT1: all exons present; cTnnT2: missing exon 4 [five amino acids]; cTnnT3: missing exon 5 [ten amino acids]; and cTnnT4: missing exons 4 and 5) [519]. The variable region in the N-terminus of cTnnT contributes to the determination of the Ca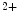 sensitivity of force development in a charge-dependent manner as well as regulation of inhibitory ATPase activity. The greater the charge, the higher the Ca
sensitivity of force development in a charge-dependent manner as well as regulation of inhibitory ATPase activity. The greater the charge, the higher the Ca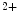 sensitivity. Calcium sensitivity of force development is associated with exon 5. All cTnnT isoforms yield the same maximal actin–tropomyosin-activated myosin ATPase activity. However, cTnnT1 or cTnnT2 that both contain information from exon 5 have a reduced ability to inhibit this ATPase activity with respect to the cTnnT3 subtype [519].
sensitivity. Calcium sensitivity of force development is associated with exon 5. All cTnnT isoforms yield the same maximal actin–tropomyosin-activated myosin ATPase activity. However, cTnnT1 or cTnnT2 that both contain information from exon 5 have a reduced ability to inhibit this ATPase activity with respect to the cTnnT3 subtype [519].
sensitivity of force development in a charge-dependent manner as well as regulation of inhibitory ATPase activity. The greater the charge, the higher the Ca sensitivity. Calcium sensitivity of force development is associated with exon 5. All cTnnT isoforms yield the same maximal actin–tropomyosin-activated myosin ATPase activity. However, cTnnT1 or cTnnT2 that both contain information from exon 5 have a reduced ability to inhibit this ATPase activity with respect to the cTnnT3 subtype [519].The cTnnT expression differs in the normal and failing adult and fetal human heart [520]. The difference is achieved by the variable inclusion of a 15- and 30-nucleotide exon in the 5′ half of the coding region. The cTnnT1 and cTnnT2 isoforms contain both 10- and 5-residue peptides encoded by the 30- and 15-nt exons (exons 4 and 5) or the peptide encoded by the 30-nt exon alone, respectively. They are expressed in the fetal heart, cTnnT2 being produced at a very low level. The cTnnT3 isoform has the five-residue peptide (hence lacking the ten-residue peptide). It is the dominant isoform in the adult heart. The peak myofibrillar ATPase activity is correlated with cTnnT4 expression [520]. The cTnnT4 subtype lacks both peptides encoded by the 30- and 15-nt exons. It is synthesized in the fetal heart and reexpressed in the failing adult heart. Mutations in the TNNT2 gene are responsible for hypertrophic cardiomyopathy. Failed hearts from patients with human idiopathic cardiomyopathy are associated with cTnnT alterations.
7.4.2 Tropomyosin
In striated myocytes, tropomyosin, an actin-binding protein, interacts with the troponin complex and actin to regulate calcium-induced muscle contraction. Tropomyosin is composed of two chains coiled around each other in a parallel fashion. It lies in the groove of actin helical filament spanning seven actin monomers (7-actin–tropomyosin–troponin module). During diastole, it blocks the myosin-binding sites on actin. Each tropomyosin molecule is coupled with a paired troponin.
A change in tropomyosin flexibility (reduced stability) decays actin filament activation by calcium and myosin and governs myocardial mechanical performance, as tropomyosin drives the cooperative activation of the myofilament, that is, facilitates the extension of actin stimulation beyond the local site where operates Ca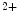 and myosin, similarly to the cooperation induced by the binding of one oxygen molecule to one hemoglobin molecule that greatly facilitates the occupancy of the remaining three oxygen-binding sites.
and myosin, similarly to the cooperation induced by the binding of one oxygen molecule to one hemoglobin molecule that greatly facilitates the occupancy of the remaining three oxygen-binding sites.
and myosin, similarly to the cooperation induced by the binding of one oxygen molecule to one hemoglobin molecule that greatly facilitates the occupancy of the remaining three oxygen-binding sites.In humans, isoforms (Tpm1–Tpm4 or Tpmα–Tpmδ) of tropomyosin are encoded by four genes (TPM1–TPM4). The three primary striated muscle isoforms include Tpm1 to Tpm3 (Tpmα–Tpmγ).
The isoforms Tpm1 and Tpm2 differ by sarcomeric performance, Tpm2 increasing sensitivity to calcium and decreasing the rate of relaxation and hence prolonging the relaxation time.
Alternative splicing multiplies the number of tropomyosin subtypes (> 40 tropomyosin isoforms) that are not functionally redundant. Each gene indeed uses alternative splicing, alternative promoters, and distinct processing to encode multiple striated and smooth muscle and nonmuscle subtypes.
Fast contracting skeletal and cardiac muscles contain more Tpm1 homodimers, whereas slow contracting muscles express more Tpm2 homodimers. The affinities of binding the Tpm1 homodimer and Tpm1–Tpm2 heterodimer to troponin-T are equivalent, whereas that of the Tpm2 homodimer to troponin-T is lower [521]. The Tpm1 homodimer exhibits higher tropomyosin–actin–myosin (S1) ATPase activity and greater Ca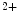 -sensitive release of troponin inhibition than the Tpm2 homodimer and Tpm1–Tpm2 heterodimer.
-sensitive release of troponin inhibition than the Tpm2 homodimer and Tpm1–Tpm2 heterodimer.
-sensitive release of troponin inhibition than the Tpm2 homodimer and Tpm1–Tpm2 heterodimer.In murine hearts, both TPM1 and TPM2 transcript levels are elevated during embryogenesis. The TPM1 mRNA/TPM2 mRNA ratio rises from 5:1 to 60:1 during the embryonic-to-adult transition [521]. Overexpression of Tpm2 reduces the maximum rate of relaxation and heightens the time needed to complete the relaxation phase and causes a concomitant decrease in TPM1 transcript levels and a preferential formation of Tpm1–Tpm2 heterodimers, but does not alter the structure of the sarcomere and the expression of any other contractile protein genes [521].
Among Tpm1 subtypes, Tpm1α (one of ten distinct products of the TPM gene) is the predominant isoform in human hearts (> 90 % [up to 98 %] in murine hearts). The substitution of the smooth muscle-specific exon 2a by exon 2b produces Tpm1κ subtype (2–5 % of total tropomyosin expressed). Exons 2a and 2b of the TPM1 gene are spliced in a mutually exclusive manner; exon 2b is the default exon in the transcript of most cell types; exon 2a is restricted to TPM1 mRNA in smooth and striated myocytes. The Tpm1κ subtype is associated with a reduced actin activation by both Ca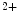 and strong cross-bridges, hence altering systolic and diastolic functions and decreasing myofilament calcium sensitivity [522].
and strong cross-bridges, hence altering systolic and diastolic functions and decreasing myofilament calcium sensitivity [522].
and strong cross-bridges, hence altering systolic and diastolic functions and decreasing myofilament calcium sensitivity [522].The heart may use the TPM1κ isoform to modulate sarcomeric performance during stress, as TPM1κ may increase flexibility of tropomyosin, thereby shortening the cooperative distance over which the thin filament is activated by calcium and myosin binding from a local stimulation [523]. Wrapped superhelically around actin filaments, tropomyosin moves smoothly and anisotropically, without any unfolding, chain separation, and localized kinks. Tropomyosin flexibility acts in the assembly and regulatory switching of actin filaments.
Among tropomyosin subtypes, the normal adult human heart expresses Tpm1α, Tpm1κ, and Tpm2 [522]. Overexpression of Tpm2 causes diastolic dysfunction and increased myofilament calcium sensitivity. The Tpm2 isoform-specific C-terminus does not cause morphological or pathological alterations, but lowers the rates of contraction and relaxation, elevates the time to peak pressure, diminishes intraventricular pressure, and heightens end-diastolic pressure [524]. It depresses maximal tension and myosin ATPase rate and reduces sensitivity of tropomyosin to calcium. Overexpression of Tpm3 raises systolic and diastolic function, but lowers myofilament calcium sensitivity without any morphological abnormalities.
Isoform switching of sarcomeric proteins is an adaptive mode of the heart in response to cardiac hypertrophy and heart failure in combination with modifications in the relative abundance and phosphorylation state of contractile and regulatory proteins. Substitution of nonmuscle Tpm1 C-terminal by muscle one affects tropomyosin affinities for troponin and actin.
Patients in a cardiomyopathy-caused end-stage heart failure have an increased Tpm1κ expression [522]. Incorporation of Tpm1κ in myofilaments actually engenders dilated cardiomyopathy.
Mutations in exon 2b of the TPM1 gene provoke familial hypertrophic cardiomyopathy (Glu62Gln [E62Q], Ala63Val [A63V], and Lys70Thr [K70T]) and dilated cardiomyopathy (Glu40Lys [E40K] and Glu54Lys [E54K]). Mutation Glu62Gln in exon 2b causes both dilated and familial hypertrophic cardiomyopathies.
7.4.3 Vinculin and Metavinculin
Vinculin is an actin-binding protein at specialized cytoskeletal-associated membrane nanodomains, as it localizes to focal adhesions and adherens junctions, in particular, focal adhesions in fibroblasts and fascia adherens of intercalated discs and costameres in cardiomyocytes. It is closely apposed to the plasma membrane at sites where actin-containing microfilaments terminate. It mediates the recruitment of numerous binding partners (talin, α-catenin, α-actinin, vasodilator-stimulated phosphoprotein [VaSP], ponsin [SorbS1],3 vinexin-α and β [SorbS3], the ARP2–ARP3 complex, paxillin, raver-14 PIP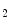 , and filamentous actin) and regulates cellular responses to tension, as it links integrin receptors to the actin cytoskeleton. Vinculin mediates cell adhesion, motility, and response to force. Metavinculin5 has, unlike vinculin, the solubility properties of an integral membrane protein [526]. It may anchor actin filaments. Metavinculin mutations are detected in some cases of dilated cardiomyopathies. Several mutations in the insert (Δ L954, A934V, and R975W) are actually associated with cardiomyopathies with improper generation of force and an impaired metavinculin–
, and filamentous actin) and regulates cellular responses to tension, as it links integrin receptors to the actin cytoskeleton. Vinculin mediates cell adhesion, motility, and response to force. Metavinculin5 has, unlike vinculin, the solubility properties of an integral membrane protein [526]. It may anchor actin filaments. Metavinculin mutations are detected in some cases of dilated cardiomyopathies. Several mutations in the insert (Δ L954, A934V, and R975W) are actually associated with cardiomyopathies with improper generation of force and an impaired metavinculin–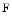 actin interaction in humans [527].
actin interaction in humans [527].
, and filamentous actin) and regulates cellular responses to tension, as it links integrin receptors to the actin cytoskeleton. Vinculin mediates cell adhesion, motility, and response to force. Metavinculin5 has, unlike vinculin, the solubility properties of an integral membrane protein [526]. It may anchor actin filaments. Metavinculin mutations are detected in some cases of dilated cardiomyopathies. Several mutations in the insert (Δ L954, A934V, and R975W) are actually associated with cardiomyopathies with improper generation of force and an impaired metavinculin–actin interaction in humans [527].In fact, metavinculin is a splice variant of vinculin. Both are scaffold proteins that localize to cellular adhesions. Metavinculin acts in mechanotransduction. Its expression level is correlated with the force exerted on cells.
Vinculin dimerizes at high concentrations in the absence of ligands and at physiological concentrations in the presence of actin and PIP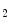 . It heterodimerizes as well as forms tetramers and higher-order oligomers with metavinculin. It also oligomerizes in the presence of different binding partners. Vinculin oligomerization is implicated in vinculin activation and hence scaffolding function, especially with
. It heterodimerizes as well as forms tetramers and higher-order oligomers with metavinculin. It also oligomerizes in the presence of different binding partners. Vinculin oligomerization is implicated in vinculin activation and hence scaffolding function, especially with 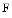 actin [527]. Vinculin can bundle
actin [527]. Vinculin can bundle 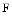 actin by homodimerization, each monomer binding to one filament, unlike α-actinin that bundles actin filaments by forming bridges between filaments. The actin bundling abilities of vinculin and metavinculin differ. Metavinculin may be able to form actin filament bundles, but may also cleave actin filaments into smaller filaments that form a meshwork [527]. Mutations in human metavinculin associated with cardiomyopathies alter the bundling activity of metavinculin.
actin by homodimerization, each monomer binding to one filament, unlike α-actinin that bundles actin filaments by forming bridges between filaments. The actin bundling abilities of vinculin and metavinculin differ. Metavinculin may be able to form actin filament bundles, but may also cleave actin filaments into smaller filaments that form a meshwork [527]. Mutations in human metavinculin associated with cardiomyopathies alter the bundling activity of metavinculin.
. It heterodimerizes as well as forms tetramers and higher-order oligomers with metavinculin. It also oligomerizes in the presence of different binding partners. Vinculin oligomerization is implicated in vinculin activation and hence scaffolding function, especially with actin [527]. Vinculin can bundle actin by homodimerization, each monomer binding to one filament, unlike α-actinin that bundles actin filaments by forming bridges between filaments. The actin bundling abilities of vinculin and metavinculin differ. Metavinculin may be able to form actin filament bundles, but may also cleave actin filaments into smaller filaments that form a meshwork [527]. Mutations in human metavinculin associated with cardiomyopathies alter the bundling activity of metavinculin.7.4.4 Perturbed Transcriptome
The transcriptome is reprogrammed and changed by a fetal program. Transcription of the gene that encodes actin-α is downregulated, whereas myosin heavy chain-β production is upregulated [515].
Expression of genes that encode proteic constituents of the cytoskeleton and extracellular matrix is deregulated. Consequently, the synthesis of collagen types 1 and 3 (encoded by the COL1A1 [Col1α1] and COL1A2 [Col1α2] and COL3A1 genes, respectively), the collagen-binding keratan sulfate proteoglycan fibromudulin (encoded by the FMOD gene), fibronectin (encoded by the FN1 gene), ubiquitous actin- and calcium-binding plastin-3 (or T-plastin encoded by the PLS3 gene), and pinin (or desmosome-associated protein encoded by the PNN gene) rises [515].
The genes that encode atrial (ANP) and brain (BNP) natriuretic peptide are the most upregulated in heart failure [515]. Transforming growth factorβ1 is also overexpressed, thereby favoring interstitial fibrosis and myocytes hypertrophy.
Transcription of genes involved in fatty acid metabolism is downregulated, whereas that of genes implicated in glucose metabolism is upregulated.
Mediators of proteolysis and stress response, such as thrombospondin-4 (encoded by the THBS4 gene), are overexpressed [515]. Antiapoptotic genes are underexpressed.
MicroRNAs that regulate gene expression posttranscriptionally contribute to the pathogenesis and evolution of cardiac diseases toward heart failure, as variations of their expression alter production of target transcripts. In particular, microRNA dysregulation is linked to cardiomyocyte hypertrophy, fibrosis, and remodeling, as well as calcium handling perturbations. As microRNAs reside in the cell as well as outside them exported and conveyed in microvesicles (exosomes, microparticles, and apoptotic bodies), they can operate as intra- and auto- (self-signaling), and juxta-, para-, and endocrine regulators (intercellular signaling between members of a given cell type as well as between diverse cell types).
Long noncoding RNAs (lncRNA), which are often expressed in a development-specific manner, constitute another category of transcripts that control gene expression. Many lncRNAs are large intergenic noncoding RNAs (lincRNAs). Long noncoding RNAs can regulate gene expression via chromatin modifications and transcription and post-transcriptional processing. In particular, HoTAiR (Hox transcript antisense RNA) from the HOXC locus prevents transcription of the HOXD locus, as it induces a repressive chromatin state by recruiting Polycomb remodeling complex PRC2 [528]. In mice, the cardiac lncRNA Braveheart (Bvht) may mediate epigenetic regulation of commitment of embryonic stem cells to cardiovascular lineages, as it interacts with suppressor of zeste homolog SuZ12, a component of Polycomb repressive complex PRC2 [529]. It functions upstream from mesoderm posterior MESP1, a master regulator of a common multipotent cardiovascular progenitor. It is required in the maintenance of cardiac fate in neonatal ventriculomyocytes. The Braveheart level in plasma of mice with induced heart failure is reduced. Long noncoding RNAs, as do certain microRNAs and cardiac transcription factors, may stimulate cellular reprogramming.
DNA methylation at CpG dinucleotides can engender transcriptional repression. Histone modifications (acetylation, methylation, and phosphorylation) also affect gene transcription. These epigenetic processes can intervene in cardiac hypertrophy and failure. A distinct DNA methylation pattern exists between patients with dilated cardiomyopathy and healthy subjects [515].
7.4.5 Heat Shock Protein-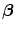 1
1
A heart failure risk locus is linked to the chromosomal region 1p36 within the HSPB7 gene that encodes 27-kDa heat shock protein, or heat shock protein-β1 [530]. A single single-nucleotide polymorphism6 (Arg83Gly) on the CLCNKA gene adjacent to HSPB7 that encodes the kidney-specific, voltage-gated chloride channel ClCK (or ClCK1) is associated with the heart failure risk connected to single-nucleotide polymorphism of the HSPB7 gene. The variant
(or ClCK1) is associated with the heart failure risk connected to single-nucleotide polymorphism of the HSPB7 gene. The variant 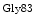 ClCK
ClCK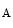 chloride channel leads to a loss of function with respect to the wild-type
chloride channel leads to a loss of function with respect to the wild-type 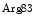 ClCK
ClCK channel.
channel.
(or ClCK1) is associated with the heart failure risk connected to single-nucleotide polymorphism of the HSPB7 gene. The variant ClCK chloride channel leads to a loss of function with respect to the wild-type ClCK channel.7.4.6 GPCR Kinase
G-protein–coupled receptor kinases phosphorylate liganded β-adrenergic receptors that then recruit β-arrestin, uncouple from Gs proteins, and internalize, thus switching off β-adrenoceptor signaling. Whereas GRK2 can mainly operate in acute regulation of β-adrenoceptor signaling, GRK5 activity that is less fast serves in long-term regulation. The Grk5 gene variant that is common in African Americans improves survival in individuals with chronic heart failure [531].7 The GRK5 variant has a higher kinase activity than that of wild-type enzyme.
7.4.7 Cardiorenal Syndrome
The cardiorenal syndrome combines cardiac and renal dysfunction that amplifies progression to failure of each organ. A vicious circle develops with activation of the renin–angiotensin axis, NO–ROS imbalance, stimulation of the sympathetic nervous system, and inflammation, all these factors synergizing with positive feedback in the cardiorenal connection [532].8 Bartter and Gitelman syndromes are autosomal recessive renal disorder. They are characterized by hypokalemia, hypomagnesemia, hypochloremia, metabolic alkalosis, and normal to low blood pressure.
The Bartter syndrome is also associated with hyperreninemia independently of extracellular volume, hyperaldosteronemia, and hypercalciuria. It results from loss-of-function mutations in the genes that encode NKCC2, K 1.1 (ROMK), ClCK
1.1 (ROMK), ClCK , and barttin in the thick ascending limb of the loop of Henle (Table 7.2).
, and barttin in the thick ascending limb of the loop of Henle (Table 7.2).
1.1 (ROMK), ClCK, and barttin in the thick ascending limb of the loop of Henle (Table 7.2).In the loop of Henle, sodium chloride is taken up apically by the combined activity of Na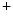 –K
–K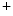 –2Cl
–2Cl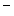 cotransporter NKCC2 and potassium channel ROMK (K
cotransporter NKCC2 and potassium channel ROMK (K 1.1). Chloride ions exit from the cell through basolateral member of the CLC family of voltage-gated chloride channels, ClCK
1.1). Chloride ions exit from the cell through basolateral member of the CLC family of voltage-gated chloride channels, ClCK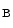 , or ClCK2 channel. Barttin, encoded by the BSND (Bartter syndrome, infantile, with sensorineural deafness) gene, is a β subunit of the ClCK
, or ClCK2 channel. Barttin, encoded by the BSND (Bartter syndrome, infantile, with sensorineural deafness) gene, is a β subunit of the ClCK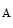 (ClCK1) and ClCK
(ClCK1) and ClCK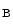 channels [533]. The former resides in both the apical and basolateral plasma membrane of the thin ascending limb of Henle’s loop. It is responsible for chloride permeability. The latter (ClCK2) lodges in the basal plasma membrane of cells of the thick ascending limb of Henle’s loop, distal convoluted tubule, connecting tubule, and intercalated cells of collecting ducts. Barttin–ClCK heteromers are involved in renal salt (NaCl) reabsorption. Barttin regulates permeation and gating of ClCK channels. In fact, barttin–ClCK heteromers enable ClCK current due to their insertion into the plasma membrane. Type-4 Bartter syndrome caused by the R8L barttin mutation results from aberrant intracellular localization [534].
channels [533]. The former resides in both the apical and basolateral plasma membrane of the thin ascending limb of Henle’s loop. It is responsible for chloride permeability. The latter (ClCK2) lodges in the basal plasma membrane of cells of the thick ascending limb of Henle’s loop, distal convoluted tubule, connecting tubule, and intercalated cells of collecting ducts. Barttin–ClCK heteromers are involved in renal salt (NaCl) reabsorption. Barttin regulates permeation and gating of ClCK channels. In fact, barttin–ClCK heteromers enable ClCK current due to their insertion into the plasma membrane. Type-4 Bartter syndrome caused by the R8L barttin mutation results from aberrant intracellular localization [534].
–K–2Cl cotransporter NKCC2 and potassium channel ROMK (K1.1). Chloride ions exit from the cell through basolateral member of the CLC family of voltage-gated chloride channels, ClCK, or ClCK2 channel. Barttin, encoded by the BSND (Bartter syndrome, infantile, with sensorineural deafness) gene, is a β subunit of the ClCK (ClCK1) and ClCK channels [533]. The former resides in both the apical and basolateral plasma membrane of the thin ascending limb of Henle’s loop. It is responsible for chloride permeability. The latter (ClCK2) lodges in the basal plasma membrane of cells of the thick ascending limb of Henle’s loop, distal convoluted tubule, connecting tubule, and intercalated cells of collecting ducts. Barttin–ClCK heteromers are involved in renal salt (NaCl) reabsorption. Barttin regulates permeation and gating of ClCK channels. In fact, barttin–ClCK heteromers enable ClCK current due to their insertion into the plasma membrane. Type-4 Bartter syndrome caused by the R8L barttin mutation results from aberrant intracellular localization [534].The Gitelman syndrome, another inherited salt-wasting disorder, causes hypocalciuria in addition to abnormal reabsorption of Na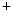 and Cl
and Cl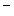 ions from the distal convoluted tubules through thiazide-sensitive Na
ions from the distal convoluted tubules through thiazide-sensitive Na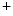 –Cl
–Cl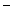 symporter (NCCT cotransporter or SLC12a3).9
symporter (NCCT cotransporter or SLC12a3).9
and Cl ions from the distal convoluted tubules through thiazide-sensitive Na–Cl symporter (NCCT cotransporter or SLC12a3).9Table 7.2
Different types of Bartter syndrome.. A symporter is also called a cotransporter
Bartter syndrome Type | Gene mutations | Ion carrier |
|---|---|---|
Type 1 | SLC12A2 | Na 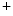 –K –K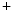 –2Cl –2Cl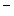 symporter (NKCC2) symporter (NKCC2) |
Type 2 | KCNJ1 | K 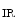 1.1 (ROMK) 1.1 (ROMK) |
Type 3 | CLCNKB | ClCK 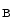 |
Type 4 | BSND | Barttin |
Type 5 | CASR | Calcium-sensing receptor |
Gitelman syndrome | SLC12A3 | Na 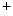 –Cl –Cl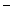 symporter (NCCT) symporter (NCCT) |
BSND Bartter syndrome, infantile, with sensorineural deafness, a ClCK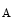 subunit
subunit
subunit7.5 Cardiac Energetics
Energy metabolism in cardiomyocytes is related to cardiac contractility. The heart has a limited fuel-storing capacity. In normal conditions, ATP is efficiently and quickly produced mainly from circulating free fatty acids and, to a lesser degree, from glucose, but fatty acids require more oxygen than glucose to produce an equivalent amount of ATP. In heart failure, the ability of mitochondria to synthesize ATP ensuring a normal cardiac function is impaired.
7.5.1 Modeling
The transition between the compensatory phase and heart failure corresponds to a reduction in the total adenine nucleotide pool (TAN) of about 30 % [535]. At given values of total adenine nucleotide and the total exchangeable phosphate pool during defective cardiac remodeling, the creatine pool attains a value that is associated with optimal ATP hydrolysis potential.
A computational model incorporates the total adenine nucleotide pool, total creatine pool (Cr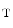 ), and total exchangeable phosphate pool (TEP) [535]:
), and total exchangeable phosphate pool (TEP) [535]:
![$$\begin{aligned} \mathrm{Cr}_{\mathrm{T}} &= V_c\alpha_{{\tt w}_c}\left(\left[\mathrm{Cr}^{\mathrm{P}}\right]_c + \left[\mathrm{Cr}\right]_c\right)\!; \nonumber\\ \mbox{TEP} &= V_c\alpha_{{\tt w}_c}\left(2\left[\mathrm{ATP}\right]_c + \left[\mathrm{ADP}\right]_c + \left[\mathrm{P}_{\mathsf i}\right]_c + \left[\mathrm{Cr}^{\mathrm{P}}\right]_c\right) \nonumber\\ &\quad\; V_m\alpha_{{\tt w}_{mims}}\left(2\left[\mathrm{ATP}\right]_{mims} + \left[\mathrm{ADP}\right]_{mims} + \left[\mathrm{P}_{\mathsf i}\right]_{mims} + \left[\mathrm{Cr}^{\mathrm{P}}\right]_{mims}\right) \nonumber\\ &\quad\; V_m\alpha_{{\tt w}_{mm}}\Big(\left[\mathrm{ATP}\right]_{mm} + \left[\mathrm{P}_{\mathsf i}\right]_{mm} + \left[\mathrm{GTP}\right]_{mm}\!\Big) \nonumber\\ \mbox{TAN} &= V_c\alpha_{{\tt w}_c}\Big(\left[\mathrm{ATP}\right]_c + \left[\mathrm{ADP}\right]_c + \left[\mathrm{AMP}\right]_c\!\Big) \nonumber\\ &\quad\; V_m\alpha_{{\tt w}_{mims}}\Big(\left[\mathrm{ATP}\right]_{mims} + \left[\mathrm{ADP}\right]_{mims} + \left[\mathrm{AMP}\right]_{mims}\!\Big) \nonumber\\ &\quad\; V_m\alpha_{{\tt w}_{mm}}\Big(\left[\mathrm{ATP}\right]_{mm} + [\mathrm{ADP}]_{mm}\Big)\end{aligned}$$](/wp-content/uploads/2016/07/A320740_1_En_7_Chapter_Equ2.gif) < div class='tao-gold-member'>
< div class='tao-gold-member'>




), and total exchangeable phosphate pool (TEP) [535]:Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
