Gene
SNP
Variants
Functional consequence
References
MTHFR
677C>T
Homocysteine mediated
ACE
rs4646994
Increased ACE activity
Ned et al. (2012)
AGT
rs11206510
M235T
LDL mediated
Feng et al. (2011)
T174M
1166A>C
SCNN1A/ENaC
T493A
Altered Na+ absorption?
Hsieh et al. (2005)
SCNN1G
rs13331086
Altered Na+/K+ plasma levels?
PAI-1
rs1799768
4G74G
Unknown
Martínez-Calatrava et al. (2007)
LR/lepr
C538T microsatellite polymorphisms in the 3′ UTR
Unknown
Akhter et al. (2012)
NOS3
rs3918226
−690C>T
Reduced NO generation?
Zhang et al. (2012)
rs1800779
−922A>G
rs3918226
−690C>T
rs1799983
Glu298Asp G>T
4.2.1 Angiotensinogen- and Angiotensin-Converting Enzyme (ACE)
The renin-angiotensin system (RAS) is classically known for its role in the regulation of blood pressure as well as of fluid and electrolyte balance. RAS is involved in the homeostasis of fluid balance and blood pressure via effects on aldosterone secretion by the adrenal cortex and direct vasoconstriction. In the RA system, angiotensinogen (AGT), the precursor of all angiotensin peptides, undergoes two enzymatic cleavages by renin and angiotensin-converting enzyme (ACE) to produce angiotensin I (Ang I) and angiotensin II (Ang II), respectively. Today, there is convincing evidence that angiotensin II and its metabolites play also an important role in the central nervous system and RAS has been implicated in aspects such as cognition, dementia, depression, anxiety, and epilepsy. RAS adversely affects stroke and cardiovascular disease. Polymorphisms in AGT, particularly the M235T and the T174M amino acid exchange, and the 1166A>C polymorphism in angiotensin II type 1 receptor have been reported as candidate risk factors for vascular disease, and the polymorphic variants M235T and T174M of the AGT gene were associated with hypertension (Mohana et al. 2012). Recently, genome-wide association data and resequencing data were reevaluated regarding the effect of AGT variants on BP using sib-pair information. It was shown that the use of sib-pair information can increase the power over using only unrelated samples by more than 40 %. These results indicate that family data are extremely informative when searching for variants underlying complex traits (Feng et al. 2011).
The insertion/deletion (I/D) variant (rs4646994) of the angiotensin I-converting enzyme (ACE) gene is one of the most studied polymorphisms in relation to blood pressure and essential hypertension in humans. However, the results are conflicting. The ACE gene contains a polymorphism characterized by either insertion (I) or deletion (D). The deletion allele occurs in approximately 55 % of the population and is associated with increased activity of the ACE enzyme. Previous research has shown that the insertion/deletion (I/D) polymorphism is a major determinant of plasma ACE activity. The polymorphism itself is thought to be a marker for a closely linked but yet unidentified sequence variant that modulates the expression of the ACE gene. It has been suggested that persons with the DD genotype (those who express, on average, the highest levels of circulating ACE) have an increased risk for myocardial infarction and coronary artery disease, particularly if they are otherwise at low risk. The evaluation of the DD allele in relation to hypertension and type 2 diabetes mellitus (T2DM) gave contradictory results. The variability of the results may be due to ethnic and geographical variations. Apparently, the effect of the polymorphism is stronger in some ethnicities. For instance, in Japan, there is good evidence that the DD genotype of ACE is a genetic risk factor for cerebro- and cardiovascular disease, especially cardiovascular events, in hypertensive patients.
A representative cohort of the United States comprising non-Hispanic white, non-Hispanic black, and Mexican Americans showed that the frequency of the I/D variant differed significantly by race/ethnicity. The D allele was associated with increased SBP but only among non-Hispanic blacks; no other significant associations were observed in any race/ethnic group (Ned et al. 2012). The reasons for these ethnic differences have yet to be elucidated.
4.2.2 Sodium Channels
Epidemiological and animal studies implicate that amiloride-sensitive epithelial sodium channels (ENaC, SCNN1) are responsible for the rate-limiting step of sodium reabsorption in the distal nephron and are important candidates in the development of hypertension. ENaC is a constitutively open channel, and factors controlling the number of active channels at the cell surface are likely to have profound effects on Na+ absorption and the amount of Na+ that is excreted in the final urine. The channel is composed of subunits which are encoded by different genes. Hsieh et al. (2005) evaluated the association of two common single-nucleotide polymorphisms (SNP) in the α-subunit of the ENac (αEnaC, SCNN1A) with ischemic cerebrovascular events (ICE). 493Arg carriers exhibited an increased adjusted odds for ICE compared to Trp/Trp carriers. Interaction analysis revealed that the relative risk was highest in women in the lowest age tertile. The effect was independent of traditional vascular risk factors. Several other genetic studies demonstrate the importance of the regulation of Na+ absorption for the maintenance of the extracellular fluid volume and blood pressure. A number of membrane-bound kinases have been identified that increase ENaC activity at the cell surface. Recently, de-ubiquitylating enzymes have been shown to increase ENaC activity in heterologous expression systems. Currently, in vivo physiological studies constitute a major challenge for the understanding of the regulation of ENaC to maintain the Na+ balance (Schild 2010).
Recently, Harrap and colleagues (Büsst et al. 2011) reported six SNPs in SCNN1G associated with systolic BP (SBP). The effect of the minor (G) allele of the SNP (rs13331086) was most important and remained significant after meta-analysis. The overall effect on SBP was 1.01 mmHg (P = 0.002) and on DBP was 0.52 mmHg (P = 0.011), after adjustment for age, sex, and body mass index. Interestingly, the rs13331086 minor allele was associated with urinary potassium excretion but not urinary sodium excretion, urinary sodium/potassium ratio, plasma sodium or potassium, or urinary aldosterone excretion (Büsst et al. 2011; Morris 2011).
4.2.3 Hypertension and the Hemostatic System
Interestingly, an association between a common SNP in the plasminogen activator inhibitor-1 gene and hypertension has been reported. The PAI-1 4G/4G (rs1799768) genotype conferred an elevated relative risk for arterial hypertension, regardless of PAI-1 levels and other hypertension-related factors. The biological and molecular mechanisms are still unresolved (Martínez-Calatrava et al. 2007). Central pressures were higher in women carrying the PAI-1 4G/4G genotype compared to females with the 5G/5G genotype. The association remained after adjustment for potentially confounding factors related to hypertension. No association of the PAI-1 genotype with blood pressure was found in men (Björck et al. 2011).
4.2.4 Hypertension and Leptin
Leptin is a hormone, mainly synthesized in adipocytes, that regulates food intake and energy expenditure of the body (see also Sect. 4.3). Rare mutations in the leptin gene cause obesity. Common polymorphisms of the leptin gene have been associated with obesity; however, recently a strong association of a microsatellite polymorphism in the 3′ untranslated region of the leptin gene with essential hypertension was found in subjects with a high BMI. The association was low in individuals with a normal BMI. Linear regression analysis showed an independent correlation of leptinemia with BMI, and a notable correlation was found between serum leptin concentration and angiotensin II. Currently, the underlying mechanism remains unknown. This association underlines the complex interactions between (patho)physiological systems (Akhter et al. 2012).
4.2.5 Nitric Oxide Synthase 3 (eNOS, NOS3)
Nitric oxide synthase 3 (NOS3) catalyzes production of NO in the endothelium and seems to play a role in cardiovascular disease (CVD). NOS3 variants were significantly associated with CHD and heart failure, and significant pharmacogenetic effects were identified for stroke and all of them cause mortality. The hazard ratio (HR) for CHD was higher in minor allele carriers at NOS3 −690C>T (rs3918226); for NOS3 −922A>G (rs1800779), a higher HR for heart failure was found in minor allele carriers. In contrast, the risk for stroke was lower in −690C>T (rs3918226) and Glu298Asp G>T (rs1799983) minor allele carriers when treated with amlodipine versus lisinopril. Data suggest that NOS3 variants may potentially provide useful clinical information with respect to treatment decisions in the future (Zhang et al. 2012).
4.3 Obesity and Type 2 Diabetes
Abnormalities designated as “secondary risk factors” for cardiovascular events as described in detail in Part II of this book generally show a strong genetic background and typically share susceptibility genes. Consequently, signaling molecules introduced above will be of importance for additional risk situations. One example is NOS3, which is a potential determinant of the severity of diabetic complications. A paradigm for tight overlap in genetic background of secondary risk factors is type 2 diabetes (T2DM) and obesity. The reader will note a significant degree of overlap in genetic risk factors and causative gene variants between the separately discussed disease states. Thus, it is important to emphasize that mutations in susceptibility genes, although causing defined cellular dysfunctions, can give rise to multiple risk phenotypes (secondary risk factors). This overlap in genetic basis of disease can create combined phenotypes representing a malign constellation of multiple “cardiometabolic” risk factors specifically regarding the “metabolic syndrome” (Reaven 1988; Mottillo et al. 2010, see also Chap. 12). Focusing on the genetic background of T2DM and obesity, a list of prominent susceptibility genes that can be considered as causative based on available knowledge of gene product functions is provided in Table 4.2.
Table 4.2
Prominent susceptibility genes in obesity
Gene | SNP | Variants | Functional consequence | References |
|---|---|---|---|---|
ADRB3 | Trp64Arg | Gjesing et al. (2008) | ||
Arg64Trp | Increased BMI | Allison et al. (1998) | ||
Arg64Trp | Increased BMI | Fujisawa et al. (1998) | ||
Arg64Trp | Increased BMI | Kurokawa et al. (2001) | ||
Arg64Trp | Increased BMI | Kurokawa et al. (2008) | ||
BDNF | Val66Met | Higher BMI | Shugart et al. (2009) | |
MC4R | V103I | Reduced risk of obesity | Zobel et al. (2009) | |
Val103Ile | Reduced risk of obesity | |||
Val103Ile | Reduced risk of obesity | |||
Val103Ile | Reduced risk of obesity | |||
NPY2R | rs12649641 | Reduced risk of obesity | ||
PCSK1 | N221D | Increased risk of obesity | Torekov et al. (2006) | |
PPARGC1B | Ala203Pro | Reduced risk of obesity | Benzinou et al. (2008) | |
FTO | rs9939609 | Andersen et al. (2005) | ||
rs9930506 | ||||
rs991121980 |
Common forms of obesity are polygenic disorders and represent a risk for a majority of chronic diseases. Consequently, obesity is considered as a trait of prominent impact on morbidity and mortality. The tight causative relation between obesity, T2DM, and cardiovascular dysfunctions is well established, and pathogenesis involves a complex interplay of genetic and environmental factors (Temelkova-Kurktschiev and Stefanov 2012). Genetic susceptibility appears to determine the individual risk to develop obesity by more than 40 % and probably even up to 70 % (Maes et al. 1997). The search for genetic variations, which, combined with environmental factors, give rise to pathologically relevant BMI levels, was initially hypothesis driven. Candidate gene studies revealed a small number of relevant genes and causative gene variants, which are robustly associated with obesity and provide the basis of severe but rare, monogenic forms of the disease such as variants of the melanocortin 4 receptor (MC4R), prohormone convertase 1/3 (PCSK1), or brain-derived nerve growth factor (BDNF) as listed in Table 4.2. These studies advanced our understanding of the molecular mechanism involved in obesogenesis but shed little light onto common obesity susceptibility. Nonetheless, with the R64Y mutation of ADRB3, one of the first genetic variation associated with both obesity and T2DM in a most likely causative manner was identified (Eisenach and Wittwer 2010), and for MC4R (Masuzaki et al. 2009), polymorphisms were found that not only give rise to rare monogenic forms of obesity but determine obesity risk also in a polygenic manner as the V103I mutation is negatively associated with BMI (Hinney and Hebebrand 2008).
4.3.1 Fat Mass and Obesity-Associated (FTO)
With the advent of large-scale GWAS, a series of common obesity-associated gene loci were identified including FTO (fat mass and obesity associated) as a repeatedly confirmed susceptibility gene with profound relevance for common obesity (Scuteri et al. 2007; Frayling et al. 2007). FTO was found associated with T2DM and subsequently uncovered as a strong determinant of BMI (Scuteri et al. 2007; Frayling et al. 2007). The FTO gene product is an about 60 kD 2-oxoglutarate-dependent nucleic acid demethylase. Despite the strong impact of FTO polymorphisms on obesity risk, the underlying molecular mechanism is still not understood. Nonetheless, a strong association was found for potentially functional SNPs in FTO and early-onset, severe obesity (Frayling et al. 2007). Recent attempts to unravel the functional coupling of FTO to cellular signaling pathways revealed a potential linkage to neuronal plasticity by interference with the NTRK2/BDNF pathway via interaction with the transcription factor C/EBPb (Rask-Andersen et al. 2011). So far, this neuronal mechanism is rather speculative and needs confirmation by further functional studies. FTO has been suggested a common risk determinant for both obesity and T2DM, and linkage of FTO expression to insulin resistance has been reported (Perry and Frayling 2008). Another interesting aspect is the dependency of the impact of FTO risk alleles on physical activity. Physical activity may specifically attenuate obesity risk in risk A-allele carriers of the FTO rs9939609 genotype (Andreasen et al. 2008).
4.3.2 Leptin and Leptin Receptors
For some prominent candidate genes, GWAS has rather excluded a role in common obesity. One example are the leptin receptors, which play a pivotal role in neuroendocrine signaling for the control of energy balance and adiposity (Münzberg and Myers 2005; Bender et al. 2011). Leptin is formed in adipose tissue and communicates the requirements for food intake and energy expenditure to the central nervous system. Leptin signaling impacts on glucose metabolism by determining insulin sensitivity in a way independent of energy balance control. Thus, leptin is considered a crucial determinant of susceptibility for obesity, T2DM, and cardiovascular risk. Common obesity is associated with disturbances in leptin signaling, most prominently involving impaired leptin sensitivity.
Leptin resistance may originate not only from defects in the leptin receptor but also from impaired downstream signal transduction or availability of the hormone at its target sites within the central nervous system. Importantly, the blood–brain barrier represents the main anatomical structure that controls the traffic of adipose tissue-derived hormone(s) to its target receptors and might therefore be involved in leptin resistance (Münzberg et al. 2005). Moreover, leptin sensitivity may be impaired by defects in signaling elements downstream of the hormone/cytokine receptor. Downregulation of leptin sensitivity is considered as a physiological process in response to altered environmental conditions (Penas-Steinhardt et al. 2011), and this adaptive process involves the protein tyrosine phosphatase 1B (PTP1B) and the suppressor of cytokine signaling (SOCS3). Specifically SOCS appear(s) to play a crucial role in both leptin and insulin resistance. The involvement and relevance of genetic factors in leptin resistance, associated with common obesity, is still unclear. Recently, common variants in the gene encoding for the downstream leptin effector JAK2 (Table 4.2), a janus-type protein kinase, were found associated with susceptibility for metabolic syndrome, suggesting a causative role of this polymorphisms in leptin and/or insulin resistance (Penas-Steinhardt et al. 2011).
4.3.3 Peroxisome Proliferator-Activated Receptor γ (PPARγ)
Another gene of which variants have been found to determine the risk for both obesity and T2DM is PPARG, which encodes a ligand-regulated transcription factor of the nuclear hormone receptor family (PPARγ) that is pivotal for adipogenesis, glucose homeostasis, and insulin resistance (Altshuler et al. 2000). Disturbances in PPARγ signaling and alterations in PPARγ-dependent gene expression have been identified as a hallmark of insulin resistance and T2DM, and a series of SNPs that associate with obesity and/or T2DM have been documented (Jeninga et al. 2009). PPARG harbors not only rare mutations potentially linked to T2DM in a causative manner but also a common variation that is clearly associated with T2DM risk. The common PPARγ P12A polymorphism (rs1801282) has been suggested to exert a profound impact on population risk while modestly affecting individual susceptibility. Interestingly, the less frequent 12A allele was found to confer a decreased risk for the disease although the proline to alanine mutation within the proteins N-terminus reduces transcriptional activity (Altshuler et al. 2000; Gouda et al. 2010). Consistently, another mutation in the N-terminal activation function-1 (AF-1) domain of PPARγ, P113Q, confers enhanced transcriptional activity, which promotes T2DM (Jeninga et al. 2009). This is interesting and unexpected, as impaired PPARγ function has repeatedly been demonstrated to promote insulin resistance due to lack of expression of PPARγ target genes required for insulin signaling. Moreover, pharmacological activation of PPARγ by thiazolidinediones has become an established strategy to increase insulin sensitivity in T2DM therapy. It may be speculated that the 12A variant, which displays modestly reduced transcriptional activity, confers a reduced T2DM risk due to a mildly suppressed function as regulator of adipogenesis. Hence, different functional modifications of PPARγ can result in similar metabolic and cardiovascular outcomes. Recently, this phenomenon was addressed and in part elucidated by resolving the crystal structure of PPARγ associated with its co-receptor RXRα and bound to DNA, ligands, and peptides (Chandra et al. 2008). Several unexpected interactions between missense mutations and specific functional domains were revealed. Furthermore, it is important to note that PPARγ has been recognized to govern also vascular remodeling and inflammatory processes, whereby the transcription factor may impact via this mechanism on atherogenesis and vascular dysfunction (Schiffrin 2005).
4.3.4 Adiponectin (AdipoQ/Acrp30)
An adipose tissue-derived signaling molecule that plays a key role in metabolic and cardiovascular pathophysiology has been identified in the mid-1990s. A 30-kDa adipokine termed adipocyte complement-related protein (Acrp30) or adiponectin (AdiopQ) represents an endogenous regulator of insulin sensitivity and glucose and lipid metabolism. Adiponectin levels are inversely correlated not only with insulin resistance and hyperlipidemia but also with the risk to develop inflammatory diseases and certain types of cancer (Ziemke and Mantzoros 2010). Thus, adiponectin signaling has emerged as a factor involved in obesity-related malignancies. The adiponectin gene is located on chromosome 3q27, within a region that determines susceptibility to T2DM. Production and secretion of the antidiabetic adipokine is governed by PPARγ and is clearly upregulated by thiazolidinediones (Combs et al. 2002). Circulating adiponectin levels are typically low in obese humans (Arita et al. 1999) and are depending on nutrition and exercise. Cellular actions of adiponectin are mediated by two different G-protein-coupled receptors (GPCRs; AdiopR1/R2) and transduced into different signaling cascades including the mTOR, JNK, and STAT3 pathways (Ziemke and Mantzoros 2010). The adipokine was reported to exert cardiovascular protection via enhanced phosphorylation of AMP-activated protein kinase, resulting in phosphorylation and increased activity of NOS3 (eNOS, see Sect. 4.2.5; Morandi et al. 2010). Interestingly, AdipoR1-mediated activation of Ca2+ entry and Ca2+/calmodulin-dependent kinase β has recently been suggested as a prominent mechanism by which the adipokine governs mitochondrial functions as well as insulin sensitivity (Iwabu et al. 2010). The adiponectin/AdipoR system may be seen as a critical signaling hub that integrates genetic and environmental/lifestyle factors into cardiovascular risk. Common as well as rare polymorphisms in the adiponectin gene have been found associated with the risk for obesity and T2DM development. Missense mutations as well as a silent mutation that is associated with decreased adiponectin levels were found to increase the risk for obesity and insulin resistance (Stumvoll et al. 2002; Yang et al. 2007b). Interestingly, a SNP (11391 G/A; rs17300539), which is associated with increased levels of the adipokine, apparently not only lacked protection against obesity-associated metabolic disturbances but even increased the risk for childhood obesity and insulin resistance (Morandi et al. 2010). Consequently, enhanced adiponectin levels may be excluded as the basis of the recently documented metabolically healthy state in humans (Karelis et al. 2004).
4.3.5 Calpain 10 and Transcription Factor 7-Like 2 (TCF7L2)
Besides PPARG two other genes were identified as potential basis of polygenic T2DM. These are calpain 10 (CAPN10) encoding for a cysteine protease (Evans et al. 2001) and TCF7L2, which encodes for a high mobility, box-containing transcription factor implicated in the control of glucose homeostasis and vascular functions. TCF7L2 was suggested to affect susceptibility to T2DM (Grant et al. 2006). This is most likely based on its role in regulation of the insulinotropic hormone glucagon like peptide-1 (GLP-1) as one of the TCF7L2 target genes. TCF7L2 functions as a nuclear receptor for β-catenin, which transduces upstream signals from the canonical Wnt pathway to transcriptional control (Prunier et al. 2004). Wnt signaling has been suggested essential for pancreatic β cell proliferation (Papadopoulou and Edlund 2005; Rulifson et al. 2007) and is linked to a variety of other transcriptional regulators (Mulholland et al. 2005). Interestingly, calpain has been proposed as a determinant of β-catenin activity, indicating potential convergence of the mechanisms by which mutations in CAPN10 and TCF7L2 determine insulin secretion and T2DM risk (Benetti et al. 2005). It is important to emphasize that among the currently identified susceptibility genes for T2DM, the majority impacts on β cell function rather than insulin sensitivity.
4.3.6 Inwardly Rectifying Potassium Channel Kir6.2 (KCNJ11) and the Sulfonylurea Receptor 1 SUR1 (ABCC8)
Two functionally well-understood gene products involved in glucose sensing and regulation of pancreatic insulin secretion are the ion channel subunits comprising the β cell ATP-sensitive K+ channel. KCNJ11 which encodes a 390 aa, two membrane spanning, pore forming channel subunit that combines with the ABC protein SUR1 to form a tetrameric K+ channel that is inhibited by intracellular ATP. This protein complex represents a paradigm metabolic sensor (Olson and Terzic 2010). Cellular ATP generation reports glucose supply of the pancreatic β cell and is translated by a membrane potential change and modulation of voltage-gated Ca2+ entry into appropriate insulin secretion. Gain-of-function mutations in KCNJ11 promote cell hyperpolarization and impair insulin secretion (Flanagan et al. 2009). Such gain-of-function mutations are the basis of rare forms of diabetes in neonates, and a common gain-of-function mutation E23K (rs5219) has been identified, which is associated with T2DM (Gloyn et al. 2003; Nielsen et al. 2003). As expected for a common gene variant, the functional consequence of this mutation on ATP sensitivity is moderate but leads to impaired glucose homeostasis and enhanced T2DM susceptibility.
4.4 Dyslipidemia
Hyper- or dyslipidemia is a trait that represents a traditional (well-recognized) risk factor for atherosclerosis and various forms of vascular disease. The causal relation between low-density lipoprotein cholesterol (LDL-C) levels and progression of atherosclerosis as well as risk for cardiovascular events is well established. It was initially substantiated by the pioneering work on the genetic basis of familial hypercholesterinemia with the identification of genetic variations in the low-density lipoprotein receptor (LDL-R) associated with elevated LDL-C and elevated risk for coronary heart disease (Lehmann et al. 1995; Brown and Goldstein 1986). Genes involved in lipid metabolism have long been considered as candidate genes to explain heritability of atherosclerosis. Interestingly, a number of candidate genes, which were identified as the basis of rare, monogenic forms of the disease, were later on found to harbor common mutations representing potential polygenic risk determinants. Recent identification of SNPs of increasing numbers of gene loci associated with blood lipid levels (Kathiresan et al. 2009) has led to the concept of complex cardiovascular disease being based on additive effects of common gene variations. Genes associated with elevated LDL-C were also found associated with cardiovascular risk specifically for myocardial infarction (Willer et al. 2008). The most prominent candidate genes with crucial impact on blood lipid levels encode for apolipoproteins and lipoprotein receptors as well as lipoprotein receptor-associated proteins such as lipoprotein receptor-related protein 6 (LRP6; Tomaszewski et al. 2009). The function and relevance of these signaling molecules are comprehensively discussed in Chap. 13. The GWAS approach for blood lipids revealed a plethora of gene loci for which a causative link to hyperlipidemia is entirely elusive and rediscovered genes with well-known functions in lipid metabolism. Moreover, this hypothesis-generating approach has indeed uncovered a series of novel players in lipid metabolism. We will discuss a few examples of established susceptibility genes and gene polymorphisms that have been suggested as potential basis of heritability of cardiovascular risk (Table 4.3).
Table 4.3
Prominent susceptibility genes in dyslipidemia
Gene | SNP | Variants | Functional consequence | References |
|---|---|---|---|---|
CELSR2, PSCR1, SORT1 | rs599839 | LDL mediated | Schunkert et al. (2010) | |
rs646776 | LDL mediated | Kathiresan et al. (2009) | ||
PCSK9 | rs11206510 | LDL mediated | Kathiresan et al. (2009) | |
MRAS | rs9818870 | Adhesion signaling | Erdmann et al. (2009) | |
LPA, LPAL2 | rs2048327 | Promotes atherothrombosis | Trégouët et al. (2009) | |
CXCL12 | rs501120 | Neointima formation after arterial injury | Schunkert et al. (2010) | |
CXCL12 | rs1746048 | Platelet activation in atherosclerotic lesions | Schunkert et al. (2010) | |
LDL-R | rs1122608 | LDL mediated | Kathiresan et al. (2009) | |
LPA | rs3798220 | Ile4399Met | Lp(a) mediated | Luke et al. (2007) |
4.4.1 Apolipoprotein E (ApoE)
This member of the apolipoprotein family is a 299aa (34 kDa) protein that is for a large part produced in hepatocytes.
The three major human isoforms apoE2/3/4 differ in aa positions 112 and 158 by polymorphisms resulting in C/C (apoE2), C/R (apoE3), or R/R (apoE4) containing proteins encoded by the APOEε2/3/4 allele. This polymorphism of APOE exhibits clear ethnic population dependence (Corbo and Scacchi 1999) with a higher frequency of the ε4 allele in Native Americans, Pygmies, and Papuans, while ε3 is prominent in societies of long agricultural history. Structural apoE diversity can be related to function and pathophysiology as the aa residues in positions 112 and 158 determine the interaction of apoE with lipoprotein particles and LDL receptors, thereby affecting blood cholesterol levels. ApoE2 confers an approximately 50-fold reduction in binding to LDL-R as compared to apoE3 and apoE4 (Raffai et al. 2001). Besides the impact on lipid homeostasis, APOE polymorphisms have been clearly demonstrated to affect signaling pathways involved in immune reactions and inflammatory responses (see Zhang et al. 2011). Polymorphism governs the ability of ApoE to modulate/suppress cytokine secretion such as TNF-α and IL-1b with E2 conferring highest and E4 lowest effectiveness. Consistently, APOE polymorphisms were found to affect mortality infectious diseases (Zhang et al. 2011). Moreover APOE genotype strongly impacts on endothelial function and signaling, and apoE4-VLDL has been shown to counteract the antiapoptotic activity of HDL in the endothelium by suppressing PI3K/Akt pathway via a mechanism involving interaction with LDL-R (DeKroon et al. 2006). Genetic variations of APOE are robustly associated with the risk for a variety of diseases that determine longevity, including not only atherosclerosis and cardiovascular events but also infections as well as neurodegenerative disorders such as Alzheimer’s disease (Kim et al. 2009). It is important to note that the role of apoE as a modulator of immune responses is certainly relevant for its significance in cardiovascular disease.
4.4.2 Sortilin 1 (SORT1)
An example for a novel gene and molecular player in lipid homeostasis more recently identified via GWAS is sortilin 1. A genome-wide screen in patients with coronary artery disease identified 1p13.3 as locus associated with both elevated LDL-C levels and cardiovascular events. Among several SNPs within this locus, all residing in a noncoding region, rs12740374, an intronic SNP in CELSR2 (cadherin, EGF LAG-seven-pass G-type receptor), was found to generate a C/EBP transcription factor interaction site and was therefore considered a causative variation (Musunuru et al. 2010). The SNP results in enhanced expression of sortilin 1 in the liver. Sortilin 1 is a multi-ligand receptor and a member of the VSP10P domain receptor family also known as neurotensin receptor 3, which is capable of interaction with apolipoproteins, lipoprotein lipase, and LDL-R-associated protein (RAP). Cellular localization of sortilin 1 includes both the cell surface and intracellular compartments, and a role of the multi-ligand receptor in endocytosis and protein trafficking has been reported (Nielsen et al. 2001). Sortilin 1 is likely to interfere with hepatic secretion of apolipoproteins (Kjolby et al. 2010) and has been demonstrated to interfere with LDL uptake into cells (Linsel-Nitschke et al. 2010). However, diverging results have been obtained in various experimental models studying the effects of genetic manipulation of sortilin 1 expression on plasma cholesterol levels (Musunuru et al. 2010; Linsel-Nitschke et al. 2010). Thus, the mechanism linking hepatic expression of this novel player in lipid homeostasis and cardiovascular risk needs further investigations.
4.4.3 Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9)
PCSK9 represents a typical example for a gene harboring both SNPs causing rare, severe mutations resulting in profound changes in LDL-C levels (>100 md/dl) but also more common variants (frequency 19 %) with mild effects (3 mg/dl) (Willer et al. 2008). Proprotein convertase subtilisin/kexin type 9 controls LDL-C plasma levels and influences the cellular fate of LDL-Rs. Representing a secreted proteinase, PCSK9 binds to LDL-Rs and targets the receptors to the lysosome promoting its degradation (Horton et al. 2007, 2009). Consistently, rare gain-of-function mutations are associated with hypercholesterinemia (Abifadel et al. 2003) and clearly increase the risk for cardiovascular events. Protective loss-of-function mutations are more common and reduce LDL-C levels. One example is the nonsynonymous R46L polymorphism (rs11591147, frequency about 3 %) which results in moderate reductions in plasma cholesterol (13 mg/dl) (Sanna et al. 2011) and a strong reduction in cardiovascular risk, which appears not sufficiently explained by the change in LDL-C (Benn et al. 2010). This discrepancy may be taken as an indication for an additional protective mechanism of the R46L mutation besides lowering plasma cholesterol (Benn et al. 2010). However, so far animal models have clearly demonstrated the impact of PCSK9 on lipid metabolism (Frank-Kamenetsky et al. 2008) while lipid-independent effects potentially related to cardioprotection are speculation. Importantly, PCSK9 has recently been demonstrated as a highly attractive target for pharmacological intervention. For example, a monoclonal antibody against PCSK9 has been tested which produces significant reductions of LDL-C in patients with familial hypercholesterinemia (Stein et al. 2012).
4.4.4 Lipoprotein Lp(a) and Apolipoprotein Apo(a)
Epidemiology has identified plasma Lp(a) levels as a predictor of cardiovascular risk (De la Peña-Díaz et al. 2000; Kamstrup 2010). Lp(a) contains the glycoprotein apo(a), which is linked by a disulfide bond to ApoB-100 (Guevara et al. 1993, see also Chap. 13). Apo(a) is structurally related to a family of serine proteases, displaying a high degree of similarity with plasminogen. The apoA gene is localized in close proximity to the plasminogen gene on chromosome 6, and its expression is strongly determined by polymorphisms in the 5′-flanking region of the gene (Suzuki et al. 1997). Moreover, structural polymorphism in apo(a) leading to isoforms with size differences has been recognized as basis of variability in Lp(a) plasma levels. The apo(a) size variation arises from the presence of a variable number of repetitive domains, which are homologous to the plasminogen kringle-4 (K4). The structural similarity to plasminogen combined with a lack of fibrinolytic protease function results in a substantial antifibrinolytic potential based on competitive interference with the plasminogen-fibrin interaction (Loscalzo et al. 1990). Apo(a) polymorphism affects both Lp(a) plasma levels and antifibrinolytic activity and thereby cardiovascular risk. Size polymorphism based on a copy number variant has been reported as a basis of variation of plasma Lp(a) levels among ethnic groups (Kraft et al. 1996a, b), and a common nonsense mutation in the K4-2 domain was identified, which results in low Lp(a) levels (Parson et al. 2004). More recently, a nonsynonymous SNP (rs3798220) encoding an Ile>Met exchange in the pseudoprotease domain was found associated with elevated circulating Lp(a) and cardiovascular risk (Luke et al. 2007). Notably, apo(a) polymorphism represents an established paradigm for overlap in the genetic basis of disturbances in lipid metabolism and hemostasis, specifically affecting the fibrinolytic system.
4.5 Arrhythmias: Structural and Electrical Remodeling
With respect to their genetic background, arrhythmias and structural diseases of the heart are positioned at some point between rare genetic variants, typically associated with high severity levels, and common forms, characterized by a rather small increment of risk. As several examples indicate, a single pathological phenotype may be linked to different genetic determinants of predisposition and may be either based on rare genetic modifications and Mendelian traits or associated with common polymorphisms combined with a significant dependency on environmental factors.
A classical structural disease of the heart is represented by primary dilated cardiomyopathy (DCM) that affects either the left or both heart ventricles showing reduced contractility and dilatation without changes in cardiac wall thickness. While secondary dilated cardiomyopathy is induced by infection, toxins, autoimmune diseases, or disorders of the endocrine system, most cases of primary dilated cardiomyopathy are due to the idiopathic form of the disease that has a clear monogenetic origin. In addition to many HCM-related gene products, dystrophin and desmin (Arbustini et al. 2000; Li et al. 1999; Taylor et al. 2007) have been identified as affected proteins, as well as actin, δ-sarcoglycan, titin, laminin, and phospholamban (Herman et al. 2012; Takeda 2003; van der Zwaag et al. 2012). In addition to rare monogenetic forms, several common gene variants were shown to affect the risk for DCM. A GWAS study genotyping 664 DCM cases in Germany identified heat shock protein B7 (HSPB7, SNP rs1739843), a protein involved in cardiac stress response, to be associated with DCM risk (Stark et al. 2010). Variations in this gene have also been found associated with sporadic and advanced heart failure (Cappola et al. 2010; Matkovich et al. 2010). Polymorphisms of presenilin-1 (SNP rs177415), a protein involved in cell apoptosis and cardiac development, were found associated with the susceptibility for DCM (Li et al. 2011). A recent GWAS study (Villard et al. 2011) identified SNP rs2234962 located within the sequence encoding for BAG family molecular chaperone regulator 3 (BAG3), a finding that is supported by a study indicating that this gene may also be affected in rare forms of DCM (Norton et al. 2011).
Cardiomyopathies as well as heart failure are typically associated with arrhythmogenesis and a certain level of electrical remodeling. Nonetheless, genetic risk for arrhythmias has largely been identified in terms of polymorphisms in genes encoding for ion channels that determine cardiac excitability and electrical properties, thus representing classical “channelopathies.” Sudden cardiac death (SCD) is a pathological phenotype responsible for 50 % of cardiovascular mortality (Kolder et al. 2012). The primary functional cause, a ventricular fibrillation (VF), may be induced by rare genetic variations associated with a clear familial heritability pattern, based on channelopathies. These forms of SCD typically occur in younger individuals. However, the majority of cases occurs in individuals above the age of 40 years and is based on VF as a consequence of coronary artery disease (Zipes and Wellens 1998). Genome-wide association studies helped to identify a number of frequent gene variants linked to these common forms of SCD. However, for many genes found in these studies, a direct causal contribution of their encoded proteins in pathophysiological pathways is not immediately evident. These genes and gene products may rather affect expression of other genes. To determine common causal molecular targets of genetic variability linked to SCD, recent studies have been investigating intermediate phenotypes of SCD risk, i.e., the effect of frequent genetic variants on specific mechanisms and processes that have a clear causal connection to SCD (Kolder et al. 2012). Some loci identified by these studies affect proteins already known to be associated with Mendelian disorders like long QT syndrome and short QT syndrome, including potassium channels. It is of note that in addition to mutations in genes encoding for ion channels, polymorphisms of nitric synthase 1 neuronal adaptor protein (NOS1AP) (Arking et al. 2006), Na/Ka ATPase β1 subunit (ATP1B1) (Lingrel 2010), and phospholamban (PLN) (Periasamy et al. 2008), key elements of myocardial function, were also found associated with VF risk.
A cardiac disorder that receives increasing attention due to its high prevalence and association with substantial health-care costs is atrial fibrillation (AF). Representing the most common form of arrhythmia with a clear familial clustering and a wide array of established, general risk factors for cardiovascular diseases such as age, gender, diabetes, and hypertension, AF has been intensively investigated in terms of its genetic basis (Rienstra et al. 2012). AF represents a paradigm for a vicious feed-forward pathogenetic process that involves structural and electrical remodeling of the atrium. Recent studies have revealed a number of common genetic variations with examples that give a hint to the molecular pathophysiology of this disorder.
4.5.1 Human Ether-a-gogo Related Gene (HERG)
VF is closely related to changes in the structure and function of HERG (KCNQ1, KCNH2, KCNE1) and/or its regulatory subunits (Amin et al. 2012; Nishio et al. 2009; Sinner et al. 2008). A polymorphism of KCNH2 (SNP rs1805123) associated with VF leads to the substitution of lysine 897 by threonine in the delayed rectifier potassium channel. The exact effect of the genetic variant on channel function is still a matter of controversy (Bezzina et al. 2003; Männikkö et al. 2010; Paavonen et al. 2003). However, a rather rare SNP (rs1805128) affecting KCNE1 (D85N) has been demonstrated to be associated with functional defects in delayed rectifiier potassium channel (IKr, Nishio et al. 2009) and a large gene survey identified the KCNE1-D85N as a possible modulator of drug-induced torsades de pointes (Kääb et al. 2012).
4.5.2 Voltage-Gated Sodium Channels (NaV)
Another intermediate phenotype involved in VF and SCD is represented by the mechanisms responsible for PR interval and QRS duration. Here, voltage-gated sodium channels (Nav1.5, SCN5A) (Shinlapawittayatorn et al. 2011) have been demonstrated to be affected by polymorphisms that are also targets of Mendelian traits leading to cardiac conduction disease (Schott et al. 1999). SCN10A, encoding Nav1.8, is the gene affected by SNP rs6795970, a single-nucleotide polymorphism responsible for changes in PR interval and QRS duration (Holm et al. 2010).
4.5.3 Paired-Like Homeodomain Transcription Factor 2 (PITX2)
GWAS studies have identified a series of genetic loci that confer susceptibility to AF. Among these loci in particular, 4q25 near the developmental transcription factor PITX2 has received particular attention (Liu et al. 2012). PITX2 is essential in embryonic development and governs development of cardiac left-right asymmetry, pulmonary myocardium, and the sleeve of cardiomyocytes that reaches into the pulmonary vein, which represents a critical origin of triggered activities leading to AF. PITX2 is a downstream target of Wnt/β-catenin signaling and, in the heart, a pivotal player in developmental signaling pathways involving factors such as Nodal, Shox2, and Tbx3 (Poelmann et al. 2008; Tessari et al. 2008; Christoffels et al. 2010).
PITX2 is the closest gene to common risk variants on chromosome 4q25 with the first identified by Gudbjartsson et al. (2007) (rs2200733 and rs2220464; see Table 4.4).
Table 4.4
Prominent susceptibility genes linked to arrhythmias: structural and electrical remodeling
Gene | SNPa | Variants | Functional consequence | References |
|---|---|---|---|---|
Dystrophin | rs128627257 | Defective myocardial force transmission | ||
Desmin | rs121913005 | Defective myocardial force transmission | Taylor et al. (2007) | |
Actin | rs104894546 | Defective myocardial force transmission | Takeda (2003) | |
δ-Sarcoglycan | rs104893869 | Defective myocardial force transmission | Takeda (2003) | |
Titin | rs199469665 | Defective myocardial force transmission | Herman et al. (2012) | |
rs199469666 | ||||
Laminin | rs80356682 | Defective myocardial force transmission | Takeda (2003) | |
rs121913570 | ||||
HSPB7 | rs10927875 | Susceptibility for DCM | ||
Presenilin-1 | rs177415 | Susceptibility for DCM | Li et al. (2011) | |
BAG3 | rs2234962 | Susceptibility for DCM | ||
NOS1AP | rs12143842 | VF risk | Arking et al. (2006) | |
rs2880058 | ||||
rs10494366 | ||||
rs12029454 | ||||
rs4657178 | ||||
rs16857031 | ||||
ATP1B1 | rs10919071 | VF risk | Lingrel (2010) | |
Phospholamban | rs12210810 | VF risk | Periasamy et al. (2008) | |
PLN | Susceptibility for DCM | van der Zwaag et al. (2012) | ||
KCNQ1 | rs12296050 | Amin et al. (2012) | ||
KCNH2 | rs2968863 | Nishio et al. (2009) | ||
KCNE1 | rs1805128 | Sinner et al. (2008) | ||
KCNH2 | rs1805123 | Lys897Thre | Associated with VF | |
KCNE1 | rs1805128 | D85N | Functional defects in IKr | Nishio et al. (2009) |
Drug-induced torsades des pointes | Kääb et al. (2012) | |||
SCN5A | rs12053903 | Susceptibility for DCM | Shinlapawittayatorn et al. (2011) | |
rs2051211 | AF risk | Schott et al. (1999) | ||
rs41312391 | SCD risk | |||
rs2200733 | ||||
SCN10A | rs6795970 | Changes in PR interval and QRS duration | Holm et al. (2010) | |
PITX2 | rs2200733 | Common risk variants on chromosome 4q25 | Gudbjartsson et al. (2007) | |
rs2220464 | ||||
KCNN3 | rs133763339 | Associated with lone AF | Ellinor et al. (2012) |
The gene is responsible for development and identity of atrial and ventricular excitability properties as well as pulmonary myocardium. This key role has been established by genetic mouse models demonstrating a potential role in AF (Wang et al. 2010a). PITX2 is considered as an inhibitor of genetic programs essential for pacemaker functions and to govern the potential for ectopic activity. Human atrium displays expression of the PITX2c isoform which was found downregulated in AF patients (Kirchhof et al. 2011). Moreover, PITX2c-deficient mice were shown to display increased sensitivity to programmed stimulation-induced AF. Importantly, lack of PITX2c was suggested to affect ion channel expression in the atrium (Chinchilla et al. 2011), including KCNQ1 and may be also other K+ channels essential for atrial repolarization.
4.5.4 Small Conductance Ca2+-Regulated Potassium Channel (KCNN3)
Cardiac electrophysiologists have for a long time ignored the presence and/or significance of small-/intermediate-type Ca2+-activated K+ channels. Only recently these channels got into the spotlight of cardiology since expression in human hearts (Xu et al. 2003) specifically in atrium has been demonstrated. Ellinor et al. (2012) recently identified a polymorphism (rs133763339) on chromosome 1q21 that is associated with lone AF. This common variation affects a member of the intermediate/small conductance Ca2+-activated K+ channel family that appears of importance for atrial action potential morphology (KCNN3). In fact, pharmacological experiments suggest that inhibition of KCNN3 prolongs atrial action potential duration and is able to terminate AF (Diness et al. 2010). Thus, KCNN3 has emerged as an attractive target for AF therapy. It is important to note that AF is of pivotal importance as a cardiovascular risk factor promoting heart failure and stroke due to disturbances in cardiac mechanics, hemodynamics, and hemostasis.
4.6 Hemostatic Dysfunctions
In the following chapter, proteins and SNPs important for hemostatic balance will be discussed regarding their contribution to the risk of vascular diseases.
The coagulation system is essential to prevent excessive bleeding and to enable wound healing after trauma. Thus, an active coagulation system is needed. However, coagulation has to be carefully balanced to avoid bleeding as well as thrombosis. This balance is guaranteed by the well-tuned interaction of the pro- and the anticoagulatory system and fibrinolysis. In case of increased or decreased concentrations, hyperactivity, or subnormal function of any one of the participating components, the fine-tuning is disturbed resulting in clinical phenotypes. During the last two decades, great effort has been made to understand the molecular genetic mechanism of the hemostatic system. The DNA sequences of all known coagulation factors have been elucidated, and many genetic alterations affecting the function of coagulation factors have been identified (Table 4.5).
Table 4.5
Genes and polymorphisms relevant for atherothrombosis and vascular diseases
Gene | SNP | Variants | Functional consequence | References |
|---|---|---|---|---|
Fibrinogen | rs1800790 | −455G>A | Increased fibrinogen plasma level | |
rs6050 | Thr331Ala | Modestly lower plasma level | ||
Prothrombin | rs1799963 | 20210G>A | Increased prothrombin plasma level | |
FV Leiden | rs 6025 | 506Arg>Gln | Impacts on inactivation of FV leads to APC resistance | |
FXII | rs1801020 | −4C>T | Association with lower FXII plasma level | |
FXIII | rs5985 | Val35Leu | Increased FXIII activity, decreased clot stability, and resistance to fibrinolysis | |
FVII | rs 6046 | Arg413Gln | Associated with lower FVII activity | |
rs510335 | −401G/T | Associated with lower FVII activity | ||
rs5742910 | 323 0/10 bp insertion | Associated with lower FVII activity | ||
rs510317 | −402G>A | Associated with higher FVIIag and FVIIc levels | ||
FVIII | rs1800291 | Asp1260Glu | Associated with FVIII levels | |
PAI-1 | rs34857375 | −675(4G/5G) | SNP modulates PAI-1 levels, influences inhibition of fibrinolysis | |
GPIa (ITGA2) | rs 1801106 | Glu534Lys | Influences GPIa activity and expression density | |
GPIIIa (ITGB3) | rs5918 | Leu59Pro | Higher number of activated platelets |
In vitro, coagulation can be initiated via two pathways: the intrinsic system comprising factor XII, prekallikrein, high-molecular-weight kininogen, and factors XI, IX, and VIII at negatively charged surfaces and the extrinsic pathway via activation of factor VII in the presence of tissue factor. Both pathways converge at a common stage involving factors X, V, and II. As a result thrombin is formed, which then converts fibrinogen (factor I) to fibrin. The cross-linking of fibrin filaments by activated factor XIII increases clot stability and its resistance to fibrinolysis. Today, we know that the intrinsic and extrinsic coagulation systems are intertwined and the in vivo activation of the coagulation cascade is complex (Fig. 4.1).
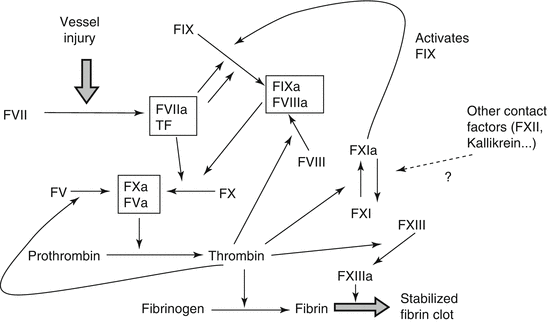
4.6.1 Coagulation Factors, Gene Polymorphisms, and Thrombotic Disease
Thrombotic events in coronary arteries are central for the development of MI. Twin and sibling studies have shown that inherited risk factors contribute significantly to the development of coronary artery disease (CAD) and ischemic stroke (Brass et al. 1996, 1992). The formation of blood clots that occlude major arteries represents the leading complication of arteriosclerosis. In the 1990s it became evident that besides mutations in coding regions of genes, polymorphisms in regulatory elements of coagulation factor genes, particularly in the promoter regions, have an important impact on blood coagulation because of their effect on the concentration of the proteins (Franco and Reitsma 2001; Blake et al. 2001). For some coagulation factors, reproducible data exist regarding the contribution of various SNPs to the variability of plasma concentration and consequently an effect on the clinical phenotype.
4.6.2 Fibrinogen (Factor I), Factor XIII, and Fibrinolysis
Fibrinogen is a 340-kDa glycoprotein consisting of three nonidentical polypeptide chains (α, β, and γ) linked by disulfide bridges. The three genes encoding fibrinogen Bβ (FGB), Aα (FGA), and γ (FGG) are clustered in a region of ∼50 kilobases (kb) on human chromosome 4. Each gene is separately transcribed and translated to produce the nascent Aα, Bβ, and γ-polypeptides, which assemble to form fibrinogen. Fibrinogen levels are subject to biological variation. They are upregulated during acute phase reactions via the activation of the IL-6 responsive elements in the promoter region of all three fibrinogen chains. High levels of fibrinogen have been associated with the development of arterial thrombosis. Prospective studies such as the Northwick Park Heart Study, the Gothenburg study, and the PROCAM study have related elevated fibrinogen levels to MI and stroke (Heinrich et al. 1994; Wilhelmsen et al. 1984). Fibrinogen levels cluster with other cardiovascular risk factors including hypertension, diabetes, smoking, and peripheral arterial disease (Lee et al. 1993; Endler and Mannhalter 2003).
Genetic factors contribute to the variability of fibrinogen plasma levels. The two most frequently studied polymorphisms are the −455G>A (rs1800790) polymorphism located in the promoter region of the fibrinogen beta chain (FGB) and the 312Thr>Ala (rs6050) polymorphism in the fibrinogen alpha chain (FGA). −455G>A is in complete linkage disequilibrium with the −148C>T polymorphism in the IL-6 responsive element and leads to increased plasma fibrinogen levels, while 312Thr>Ala is associated with modestly lower plasma fibrinogen levels. The FGA rs6050 single-nucleotide polymorphism (SNP) indicates a decreased risk, whereas the FGB −455G>A SNP seems to increase the risk of stroke. The risk of myocardial infarction does not seem to be altered by either SNP (Siegerink et al. 2009). Interestingly, an association between poststroke mortality and the 312Thr>Ala polymorphism in patients with atrial fibrillation had been observed by Carter et al. (1999). Thus, although the general risk for MI or stroke apparently is not significantly affected by fibrinogen polymorphisms, they may play a role in subgroups of patients.
Currently, the role of fibrinogen polymorphisms in thrombotic disease has still not been definitely proven. The available results suggest that plasma fibrinogen levels could be more important as risk factors for ischemic stroke than for myocardial infarction.
Fibrinogen is cleaved by thrombin leading to the generation of fibrin monomers which have to polymerize and be cross-linked by factor XIII. This cross-linking of the fibrin polymers is essential for clot stability and its resistance to fibrinolysis (Siebenlist et al. 2001). Fibrin structure and stability have been linked to thrombotic diseases. Biochemical studies of fibrinogens have examined the interactions that mediate the conversion of soluble fibrinogen to the insoluble fibrin network. These studies show that fibrin structure modulates the enzymatic lysis of the fibrin network. Thus, the molecular mechanisms that control structure also control stability. FXIII which catalyzes the formation of covalent ε(γ-glutamyl)-lysyl bonds between the γ and α chains of fibrin plays an important role in this process. Factor XIII circulates in plasma as a heterotetramer consisting of two catalytic A-subunits and two carrier protein B-subunits (A2B2). Monocytes and megakaryocytes synthesize FXIII, and it has also been identified in platelets. In the F13 gene, a common G>T polymorphism at nt163 (rs5985, c.103G>T), leading to a valine (Val) to leucine (Leu) substitution at amino acid position 35 (nomenclature according to HGVS, three amino acids away from the thrombin activation site in exon 2 of the FXIII A-subunit gene), was described (Mikkola et al. 1994). Due to the close proximity of the thrombin activation site, this polymorphism is thought to impair FXIII activation and contribute to the pathogenesis of thrombotic disorders (Kohler and Grant 1999). The Val35Leu polymorphism leads to faster activation of FXIII. In vitro, the 35Leu allele confers an increased catalytic activity to FXIII but decreases clot stability and resistance to fibrinolysis by alterations of the clot structure (Ariens et al. 2000). A lower prevalence of the 35Leu allele has been found in subjects with MI or deep vein thrombosis compared to healthy controls (Corral et al. 2000; Bereczky and Muszbek 2011). In spite of some positive reports, the importance of the FXIII 35Leu genetic variant for premature coronary artery disease (CAD) and thrombotic events remains controversial. The evaluation of its effect on long-term clinical outcome defined as a composite of cardiovascular death, recurrent MI, and revascularization indicated no association of FXIII 35Leu with premature CAD or clinical outcome. However, carriage of factor XIII 35Leu leads to a stepwise decrease in the rate of fibrinolysis with a significant gene dose effect in patients (Silvain et al. 2011). The role of the FXIII Val35Leu polymorphism in ischemic stroke is still unclear. Our group found no differences in genotype distribution between patients with ischemic stroke and healthy controls. This suggests that an association of the FXIII Val35Leu polymorphism with a decreased risk of ischemic stroke or an increased risk of intracerebral hemorrhage is unlikely (Endler et al. 2003). The results were recently confirmed by Shemirani et al. (2010) who also found no association between the risk of acute ischemic stroke and FXIIIA 35Leu carriership in either gender. Thus, while there is increasing evidence that the 35Leu allele protects from MI, its role in the development of stroke needs to be clarified.
In mammalian blood the fibrinolytic system is essential for the dissolution of blood clots and the maintenance of a patent vascular system. Abnormalities in the fibrinolytic system have been implicated in the pathogenesis of atherosclerotic diseases such as myocardial infarction and stroke (Babu et al. 2012). Fibrinolysis is initiated by specific interactions between its main components, the inactive proenzyme plasminogen, which is converted to the active enzyme plasmin, which degrades fibrin. Two plasminogen activators exist in blood: tissue-type plasminogen activator (tPA) and urokinase. Activation of plasminogen by tPA is enhanced in the presence of fibrin or at the endothelial cell surface. Inhibition of fibrinolysis occurs either at the level of plasminogen activation by a plasminogen activator inhibitor or at the level of plasmin by a2-antiplasmin.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


