Fungal Pulmonary Infections
INTRODUCTION
• Infections of the lung with both opportunistic and endemic fungi are increasingly common and are a result of the increasing population of immunocompromised hosts, due to AIDS, chemotherapy, organ transplantation, and chronic steroid use. Patients with neutropenia or lymphocytic deficiencies are predisposed to mycotic infection and pulmonary involvement is the most common form of invasive fungal disease.
• This chapter examines the clinical presentation, diagnostic approach, and treatment of the most common fungal infections of the lung.
• There are three primary forms of fungi that infect the lungs:
 Yeasts, which appear as budding forms, include species of Candida, Cryptococcus species, and Pneumocystis jirovecii.
Yeasts, which appear as budding forms, include species of Candida, Cryptococcus species, and Pneumocystis jirovecii.
 Molds, which appear as hyphae, include Aspergillus and the zygomycota—Mucorales, Fusarium, and Scedosporium.
Molds, which appear as hyphae, include Aspergillus and the zygomycota—Mucorales, Fusarium, and Scedosporium.
 Dimorphic fungi appear as both budding forms in tissue and hyphae in cultures incubated at 25°C, and include Histoplasma, Blastomyces, and Coccidioides.
Dimorphic fungi appear as both budding forms in tissue and hyphae in cultures incubated at 25°C, and include Histoplasma, Blastomyces, and Coccidioides.
• This review of fungal pulmonary infections is not intended to be exhaustive; rather, the most common and emerging fungal infections will be reviewed.
ASPERGILLOSIS
• Aspergillus is a ubiquitous soil-dwelling organism with a worldwide distribution that is found in dust, compost, foods, spices, and rotted plants. Inhalation of the spores is common, but disease is rare.
• Aspergillus fumigatus is the most commonly implicated organism but any of the Aspergillus species, including A. flavus, can cause disease.
• Risk factors for developing infection include neutropenia, prolonged steroid use, AIDS, use of chemotherapy, hematopoietic or solid-organ transplantation, chronic granulomatous disease, and pre-existing lung disease.1,2
• The Aspergillus species cause a spectrum of clinical syndromes in the lung.1,2
 Aspergilloma typifies the saprophytic processes of the lung.
Aspergilloma typifies the saprophytic processes of the lung.
 Pulmonary aspergillosis may be categorized as saprophytic, invasive, or allergic.
Pulmonary aspergillosis may be categorized as saprophytic, invasive, or allergic.
 Invasive pulmonary aspergillosis (IPA) develops as a bronchopneumonia or invasive sinusitis, and may be complicated by hemorrhage, invasion of adjacent structures, or dissemination to extrathoracic organs.
Invasive pulmonary aspergillosis (IPA) develops as a bronchopneumonia or invasive sinusitis, and may be complicated by hemorrhage, invasion of adjacent structures, or dissemination to extrathoracic organs.
 Chronic necrotizing aspergillosis (CNA) is a more indolent form of invasive infection.
Chronic necrotizing aspergillosis (CNA) is a more indolent form of invasive infection.
 Allergic conditions include allergic bronchopulmonary aspergillosis (ABPA).
Allergic conditions include allergic bronchopulmonary aspergillosis (ABPA).
Aspergilloma:
General Principles
• An aspergilloma is the most common form of pulmonary aspergillosis.1
• Also called a fungus ball, the aspergilloma is composed of fungal hyphae, mucus, inflammatory cells, fibrin, and tissue debris.
• It usually occurs in patients with pre-existing cavitary lung disease. Rare cases of de novo aspergilloma in patients without pre-existing cavitary lung disease have been reported.1,3
• Risk factors include TB, sarcoidosis, neoplasm, cystic fibrosis, or severe emphysema.
Diagnosis
• Most patients with aspergilloma have few clinical symptoms.
• When symptoms develop, hemoptysis is the most common and is usually mild but may occasionally be severe. Bleeding usually occurs from bronchial vessels lining the lung cavity.1,3
• Patients may also experience chest pain, dyspnea, malaise, and fever.
Diagnostic Testing
Laboratories
• Sputum cultures for Aspergillus are positive in only 50% of cases due to the limited communication between the cavity and the bronchial tree.1
• Serum IgG antibodies are very sensitive but may be negative in patients on corticosteroid therapy.1,3
• Aspergillus antigen has been found in the bronchoalveolar lavage (BAL) fluid of patients with aspergilloma, but the diagnostic utility is variable.
Imaging
• On CXR, aspergillomas classically appear as an upper lobe intracavitary mass surrounded by a radiolucent crescent, known as a crescent sign.
• However, these radiographic findings may also appear in other conditions, including neoplasm, abscess, cystic echinococcosis, and granulomatosis with polyangiitis.
Treatment
• Treatment of aspergilloma is usually considered only when patients become symptomatic and there is no consensus on treatment of choice.1,3,4
• Inhaled, intracavitary and endobronchial instillations of antifungal agents have been attempted, and have failed to demonstrate benefit in the clinical course, morbidity, or mortality.
• Oral itraconazole therapy has been shown to reduce the size of aspergillomas.
• Administration of amphotericin B percutaneously into the aspergilloma may be effective in patients with massive hemoptysis, with resolution of hemoptysis in several days.
• Bronchial artery embolization should be considered as a temporizing measure in patients with life-threatening hemoptysis.
• Surgical resection should be considered in patients who have recurrent hemoptysis, but resection is often limited by the underlying lung disease.
Invasive Pulmonary Aspergillosis:
General Principles
• IPA is characterized by direct vascular invasion by the fungus, often with dissemination to other organs.
• It is a rapidly progressive, frequently fatal disease that occurs primarily in immunocompromised hosts.
• Aspergillus has a propensity for vascular invasion, resulting in vascular thrombosis, infarction, and tissue necrosis. It can also invade into adjacent structures, including the intercostal muscles, ribs, and pericardium.
• Risk factors for IPA include prolonged neutropenia or neutrophil dysfunction, hematopoietic stem cell and solid organ transplantation, prolonged and high-dose corticosteroid therapy, hematologic malignancy, advanced AIDS, and cytotoxic therapy.1,2
Diagnosis
• IPA should be suspected in immunocompromised hosts with high fevers that do not respond to treatment with broad-spectrum antibiotics.
• The most common clinical manifestations include pleuritic pain, hemoptysis, pulmonary hemorrhage, and cavitation.1,2
• Diagnosis of IPA is difficult due to its nonspecific clinical manifestations, and a high index of suspicion must be present in patients with risk factors. As a result, diagnostic criteria have been proposed, incorporating histologic, microbiologic, and antigenic findings. The criteria are summarized in Table 15-1.1
TABLE 15-1 DIAGNOSTIC CRITERIA FOR THE DIAGNOSIS OF IPA

Diagnostic Testing
• Isolation of Aspergillus from the sputum has a positive predictive value of 80–90% in immunosuppressed patients; however, negative sputum studies occur in 70% of patients with confirmed IPA.1
• Serum and BAL fluid testing for galactomannan antigen is available but the operating characteristics of such testing are variable, with specificity being better than sensitivity.2,5–7 However, some medications, including β-lactam antibiotics, may be associated with a false-positive result, and antifungals with activity against Aspergillus may be associated with a false-negative result. Serum polymerase chain reaction (PCR) testing may also be available in the near future.2,8
• CXR findings are nonspecific, and may show patchy infiltrates or nodular opacities.1
• The imaging modality of choice is high-resolution CT scanning, which may reveal multiple nodules and an area of infiltrate surrounded by a rim of air, representing pulmonary necrosis, known as the halo sign.1,3
• Histopathologic diagnosis of lung tissue, obtained from thoracoscopic or open lung biopsy, remains the gold standard.
• Histologic examination demonstrates vascular invasion by septate, acute-angle–branching hyphae.
• Bronchoscopy with BAL is particularly helpful in patients with diffuse lung involvement, and has a sensitivity and specificity of 50% and 97%, respectively.1
Treatment
• Despite the introduction of new antifungal agents, the mortality of IPA remains high. Therapy should be initiated as soon as there is clinical suspicion of IPA.
• The current treatment of choice is voriconazole at 6 mg/kg IV bid on day 1, followed by 4 mg/kg IV daily for an additional 6 days. After 6 days of IV therapy, switching to voriconazole 200 mg PO bid can be considered.1,2,4
• Amphotericin B is also a first-line therapy, recommended dose 1–1.5 mg/kg/d; however, it is associated with nephrotoxicity and electrolyte disturbances. In patients who cannot tolerate amphotericin B due to renal dysfunction, the less toxic lipid formulations may be tried.1,2,4
• Optimal duration of therapy is unknown, but patients may be switched to oral itraconazole for at least 6–12 weeks only after immunosuppression has ended and there has been resolution of the lesions.
• Posaconazole, caspofungin, micafungin, and anidulafungin may also be used as salvage therapy in patients who do not respond to standard antifungal therapy.2,4
• Combination therapy has not been shown to be more effective than single-agent therapy.
• Surgical resection of infected tissue should be considered when there is massive hemoptysis or localized focus of infection, especially if further immunosuppression is planned.
Chronic Necrotizing Aspergillosis:
General Principles
• CNA is a more indolent form of invasive infection characterized by local invasion of lung tissue. Unlike IPA, CNA is slowly progressive and vascular invasion or extrathoracic dissemination is unusual.
• CNA most commonly affects elderly patients with underlying chronic lung disease, particularly chronic obstructive pulmonary disease (COPD). Other risk factors include pulmonary TB, prior lung resection, prior radiation therapy, pneumoconiosis, and cystic fibrosis.1,3
• Patients with mild immunosuppression due to diabetes mellitus, chronic liver disease, corticosteroid therapy, alcohol abuse, and connective tissue disease, are also at increased risk.1,3
Diagnosis
• Patients with CNA typically present with constitutional symptoms, including fever, malaise, fatigue, and weight loss of 1–6 months’ duration.
• Patients may also have chronic productive cough and hemoptysis of varying severity.
• Occasionally, patients are asymptomatic and the diagnosis is made incidentally.
• Patients must meet all of the following criteria in order for the diagnosis to be made1:
 Chronic pulmonary or systemic symptoms, including at least one of: weight loss, productive cough, or hemoptysis.
Chronic pulmonary or systemic symptoms, including at least one of: weight loss, productive cough, or hemoptysis.
 No overt immunocompromising conditions.
No overt immunocompromising conditions.
 Cavitary pulmonary lesion with evidence of paracavitary infiltrates, new cavitary formation, or expansion of cavity size over time.
Cavitary pulmonary lesion with evidence of paracavitary infiltrates, new cavitary formation, or expansion of cavity size over time.
 Elevated levels of inflammatory markers.
Elevated levels of inflammatory markers.
 Isolation of Aspergillus from pulmonary or pleural cavity, or positive serum Aspergillus precipitins test.
Isolation of Aspergillus from pulmonary or pleural cavity, or positive serum Aspergillus precipitins test.
 Exclusion of other pulmonary pathogens.
Exclusion of other pulmonary pathogens.
Diagnostic Testing
• Serum IgG antibodies to A. fumigatus are variably positive during the course of the disease.
• CXR and chest CT show consolidation, pleural thickening, and cavitary lesions in the upper lobes. Aspergilloma may be seen in half of patients. The radiographic findings may progress over weeks to months.
• As with IPA, diagnosis requires the histologic demonstration of tissue invasion by the fungus as well as the growth of Aspergillus on culture. However, transbronchial biopsy specimens and percutaneous aspirates have poor yield, and thoracoscopic or open lung biopsy is rarely performed, resulting in delayed diagnosis.1
Treatment
• Voriconazole 200 mg PO bid is the current primary therapy for CNA, but both itraconazole 400 mg daily and amphotericin B 0.5–1 mg/kg daily are considered first-line therapies as well. The duration of therapy is usually prolonged but optimal duration and criteria for discontinuing therapy are currently unclear.1,4
• Markers for response to therapy include weight gain, improved energy levels, improved pulmonary symptoms, decreasing inflammatory markers, decreasing total serum IgE levels, and reduction in cavity size.3
• Surgical resection can be considered in a very limited population, including young healthy patients with limited disease and good pulmonary reserve, patients not tolerating antifungal therapy, and patients with residual localized disease despite antifungal therapy.
Allergic Bronchopulmonary Aspergillosis
General Principles
• Unlike the previously discussed forms of pulmonary aspergillosis, ABPA is a hypersensitivity reaction rather than a true infection.
• The pathogenesis is not completely understood, but specific IgE-mediated type I hypersensitivity reactions, specific IgG-mediated type III hypersensitivity reactions, and abnormal T-lymphocyte cellular immune responses have all been implicated.9
• It most commonly occurs in patients with asthma or cystic fibrosis.9
Diagnosis
• Patients with ABPA usually present with episodic wheezing, occasional productive cough, fever, and chest pain.
• Patients may also complain of expectoration of thick brown plugs, which are Aspergillus-laden mucoid bronchial casts.
• Patients in whom ABPA is suspected should undergo a specific diagnostic evaluation, outlined below in Figure 15-1.9
• Patients with ABPA can be classified as those with central bronchiectasis (ABPA-CB) and those without (ABPA-seropositive).
• The minimal diagnostic criteria for ABPA-CB include asthma, skin reactivity to Aspergillus antigens, serum IgE >1000 IU/mL, elevated serum A. fumigatus-specific IgG and IgE, and central bronchiectasis.9,10
• The minimal diagnostic criteria for ABPA-seropositive include asthma, skin reactivity to Aspergillus antigens, serum IgE >1000 IU/mL, history of transient pulmonary infiltrates, and elevated serum A. fumigatus-specific IgG and IgE.9,10
Diagnostic Testing
• Early detection of ABPA prior to the development of irreversible bronchiectasis can help to minimize the severity of disease. A delay in diagnosis may result in irreversible pulmonary damage.
• Serum IgE and IgG levels are useful in diagnosing ABPA.
• The CXR may be clear or show transient migratory infiltrates in the upper lobes that occur during acute exacerbations. The ring sign and tram lines are radiographic findings that signify the thickened and inflamed bronchi.
• The chest CT scan may show mucoid impaction, bronchial thickening, or bronchiectasis, usually in a central upper lobe distribution.
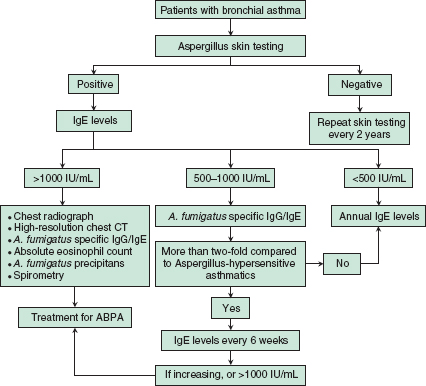
FIGURE 15-1. Diagnostic evaluation for allergic bronchopulmonary aspergillosis (ABPA). (Adapted from Agarwal R. Allergic bronchopulmonary aspergillosis. Chest. 2009;135:805–26.)
Treatment
• Glucocorticoids are the mainstay in treatment of ABPA.4,9
• Although there is little data to guide the dosing or duration of therapy, lower doses of glucocorticoids have been associated with more frequent relapses.
• Current recommendations are to treat with prednisone 0.5 mg/kg/d PO daily for2 weeks, followed by a slow taper over 3–6 months.
• Itraconazole, 200 mg bid, has been used as a steroid-sparing agent.4
• Total serum IgE can be used as a marker of disease activity, and should be checked6–8 weeks after starting glucocorticoid therapy, then every 8 weeks for 1 year after that to determine a baseline serum IgE level.
• Although oral itraconazole has been shown to reduce IgE levels, it has not been demonstrated to improve lung function.
BLASTOMYCOSIS
General Principles
• Blastomycosis is a fungal infection caused by the dimorphic fungus Blastomyces dermatitides, which exists as a mold in nature and converts to a broad-based budding yeast at body temperature.
• It appears to be most commonly found in moist soil close to bodies of fresh water, and is traditionally thought to be endemic in the Ohio and Mississippi river valleys, but the epidemiology is not well delineated.11–13
• Like many of the other fungal pulmonary infections, infection most commonly occurs when the conidia are aerosolized and inhaled, although cases of transmission by the bite of an infected animal have also been documented.11,13
• Hosts with impaired cell-mediated immunity are at greater risk of developing disseminated disease.
• Unlike histoplasmosis, blastomycosis does not appear to be a self-limited disease, and must be treated once diagnosis is made as mortality rates approach 60% in untreated disease.
Diagnosis
Clinical Presentation
History
• Clinical presentations of blastomycosis infection are highly variable, and range from mild to fatal disseminated disease.11–13
• Most primary infections are asymptomatic or mild and resolve without treatment. Of those who are symptomatic, the illness most frequently manifests as a mild, self-limited pulmonary infection.12
• Patients who are ill enough to come to medical attention present with cough, fever, night sweats, weight loss, chest pain, dyspnea, myalgias, and hemoptysis. Patients with widely disseminated or miliary disease may present with acute respiratory distress syndrome (ARDS).
• Other common sites of dissemination include the bone, joints, genitourinary system, and central nervous system (CNS), but blastomycosis can involve virtually any organ.
Physical Examination
• Approximately half of patients with disseminated disease have either ulcerative or verrucous skin lesions, which are tender and frequently confused for panniculitis.11–13
• Verrucous lesions have a raised, irregular border over an abscess in the SC tissue, with crusting and drainage.
• Ulcerative lesions have sharp, heaped-up borders and the base contains exudate.
Diagnostic Testing
• Diagnosing blastomycosis remains difficult due to the nonspecific symptoms, and delays in diagnosis are common. Blastomycosis is often not considered until the patient has failed to respond to standard treatment for bacterial infections and becomes gravely ill.
• Culture of sputum, BAL fluid, tracheal secretions, or of the skin lesions is the definitive diagnostic test but it may take up to 4–5 weeks for the organism to be isolated from culture.
• Microscopic examination of sputum, aspirate, or tissue shows the classic broad-based budding yeast, and can lead to more rapid diagnosis.
• A urine antigen assay has a reported sensitivity of 93% and a specificity of 79% but histoplasmosis, paracoccidioidomycosis, and other fungal infections may result in false positives.11,12
• Serologic testing has traditionally not been useful but advances in technology made testing more sensitive and specific.
• CXR frequently demonstrates alveolar or mass-like infiltrate, often with cavitation, and can sometimes be mistaken for malignancy.
Treatment
• In patients with non-CNS, non–life-threatening disease, itraconazole 200 mg PO daily for 6–12 months is recommended; this may be increased to 200 mg PO bid for patients not responding to therapy.4,11–13
• In patients with severe, life-threatening disease, amphotericin B 0.7–1.0 mg/kg daily for 1–2 weeks remains the mainstay of therapy. Lipid formulations may be used at 3–5 mg/kg daily.4 Once satisfactory clinical response has been documented, patients may be switched to itraconazole for a total of 1 year of therapy.
• CNS disease requires amphotericin B for 4–6 weeks prior to being switched to itraconazole.
• In immunosuppressed patients, lifelong suppressive therapy is recommended if the immunosuppression cannot be corrected.
CANDIDIASIS
General Principles
• Candida is a dimorphic fungus that is a common human saprophyte, found normally in the gastrointestinal (GI) tract and other mucocutaneous regions.
• Although Candida albicans is the major pathogenic species, other Candida organisms can also cause infection, such as C. parapsilosis and C. glabrata.14,15
• Isolated pulmonary candidiasis is rare and pulmonary involvement is usually secondary to disseminated candidiasis.16 When primary pulmonary candidiasis occurs, it is usually the result of aspiration from the oropharynx.
• Risk factors for developing disseminated candidiasis include intensive care unit hospitalization, hematologic and solid organ malignancy, hematopoietic stem cell and solid organ transplantation, use of central venous catheters, antibiotic therapy, total parenteral nutrition, and prior fungal colonization.14,15
Diagnosis
• Symptoms of pulmonary candidiasis are nonspecific, and include cough productive of purulent sputum and hemoptysis.
• Signs of disseminated candidiasis include skin lesions, endophthalmitis, and high fevers not responsive to broad-spectrum antibiotic therapy.
Diagnostic Testing
• Because the oropharynx is colonized with Candida, isolation of Candida spp. from the sputum has modest operating characteristics and is nondiagnostic.14,17,18
• CXR may show a focal or diffuse patchy infiltrate with primary disease. A military nodular pattern is more common in disseminated disease.
• Definitive diagnosis of pulmonary candidiasis requires obtaining tissue via bronchoscopic or open lung biopsy and histologic documentation of invasion of the bronchi or lungs.
• Isolation of the organism from bronchial washings or BAL fluid is more representative but may also be contaminated/colonized.19 In the correct clinical setting, repeated heavy growth of the organism from bronchial washings or BAL fluid may be used to diagnose candidiasis.
Treatment
• Fluconazole is the current treatment of choice in sensitive organisms at doses of 400 mg daily for 2–4 weeks. When using fluconazole for the treatment of candidiasis, speciation and susceptibility information is important as drug resistance is becoming common.14,15,20
• In patients who have failed fluconazole, IV amphotericin B in doses of 0.5–1.5 mg/kg daily may be used. Lipid preparations of amphotericin B may be used if amphotericin B is not tolerated.
• The echinocandins have also been shown to be effective, and have shown promise as a potential first-line agent for invasive candidiasis.
COCCIDIOIDOMYCOSIS
General Principles
• Coccidioides immitis is a dimorphic fungus that is endemic to the southwestern region of the United States.
• Infection occurs with the inhalation of arthroconidia, and has five primary manifestations: acute pneumonia, chronic progressive pneumonia, pulmonary nodules and cavities, extrapulmonary nonmeningeal disease, and meningitis.21,22
• AIDS, organ transplantation, use of tumor necrosis factor-α (TNF-α) antagonists, malignancy, and pregnancy are risk factors for acquiring coccidioidomycosis. Those of Filipino or African-American descent are at greater risk for developing disseminated disease.21–23
Diagnosis
• Approximately 60% of patients who become infected with Coccidioides are asymptomatic, and of those who are symptomatic, the vast majority develops acute pneumonia.21,24
• Symptoms develop 1–3 weeks after exposure, and are nonspecific, including fever, sore throat, cough, fatigue, and pleuritic chest pain; rarely, patients present with ARDS.
• It is sometimes associated with rashes, most classically erythema nodosum and erythema multiforme in a necklace pattern around the neck.22
• Approximately 1–4% of patients fail to recover from the acute pneumonia, and continue to have chronic progressive pneumonia, characterized by fever, weight loss, and productive cough.21–23
• Over the course of infection, the pneumonic process may consolidate to form a pulmonary nodule or a cavity. Patients may present with hemoptysis, cough, fever, night sweats, weight loss, and localized chest wall pain, but the majority of patients are asymptomatic.
• In ≤5% of immunocompetent hosts, coccidioidal infection evolves into disseminated disease.22 The most common sites of dissemination include the skin, bones, joints, lymph nodes, and meninges. Dissemination is often not diagnosed until several months after the initial pulmonary symptoms. Patients with meningitis often present with headache, mental status changes, and neurologic deficits, most commonly cranial nerve palsies. Prognosis, even in immunocompetent hosts, is grave if not diagnosed and treated promptly.
Diagnostic Testing
Laboratories
• Diagnosis of coccidioidomycosis can be made in three ways: identification of the spherules in a cytology or biopsy specimen, isolation of the organism from culture, or a serologic test that is positive for Coccidioides.22,23
• Identifying spherules in sputum, BAL fluid, or other body fluid is indicative of disease, as Coccidioides is not a colonizing organism.22
• Active cultures of pathogenic Coccidioides pose an infection risk for laboratory personnel.
• An IgM antibody to coccidioidin may be measured with several different methods, and IgG complement fixing antibody can be quantified. The IgG is present at lower titers in early disease, and higher in disseminated disease. A titer ≥1:16 should prompt investigation for disseminated disease.21,22 Negative serology does not rule out coccidiomycosis due to less than perfect sensitivity. Titers should also be quantified in the cerebrospinal fluid (CSF) if meningitis is suspected.
Imaging
• CXRs in acute pneumonia most frequently reveal dense, upper lobe infiltrates; hilar and mediastinal lymphadenopathies are not uncommon. In chronic pneumonia, CXRs may show persistent infiltrates, fibrosis, cavitation, hilar lymphadenopathy, and pleural changes.
• In cavitary disease, CXR classically shows a thin-walled lesion without an air–fluid level. In 90% of patients the cavities are solitary and in 70% they are usually 2–4 cm in size.23
• In disseminated disease, CXR most commonly shows diffuse reticulonodular infiltrates, although miliary infiltrates, nodules, and cavities may be seen.
Treatment
• In immunocompetent patients with mild symptoms, antifungal therapy is not indicated.4
• In immunocompetent patients with moderate pulmonary disease, an oral azole may be used, most commonly fluconazole 400 mg daily or itraconazole 400–600 mg daily.4,21,22,24
• In patients with disseminated disease or severe pulmonary disease, IV amphotericin B 0.5–1.5 mg/kg/d or its lipid formulations should be used until the patient has clinically stabilized, at which time they may be switched to maintenance therapy with an oral azole for at least 12–18 months.4,21,22,24
• Fluconazole is not efficacious in bone and joint disease.
• If the patient is immunosuppressed, they may require lifelong suppressive therapy with an oral azole.4
• In patients who are asymptomatic but have nodules or cavitary lesions, CXRs to monitor the size of the lesions are indicated; in the event that the patient becomes symptomatic, surgical resection may be indicated.
CRYPTOCOCCOSIS
General Principles
• Cryptococcosis is an opportunistic infection caused by Cryptococcus neoformans or C. gatti, an encapsulated, budding yeast.
• C. neoformans is found worldwide, and are especially abundant in soil that is contaminated with pigeon droppings.
• Cryptococcosis occurs when fungal cells are inhaled, and in the absence of a robust immune response, the fungus can disseminate to CNS, resulting in meningitis or meningoencephalitis.
• Risk factors for developing clinically significant disease include HIV infection, diabetes mellitus, chronic liver disease, hematologic malignancies, solid organ or hematopoietic stem cell transplantation, corticosteroid therapy, use of TNF-α antagonists, sarcoidosis, and connective tissue diseases.25–28
Diagnosis
• The majority of immunocompetent hosts infected by Cryptococcus are asymptomatic. Those with symptoms frequently complain of nonproductive cough, fever, malaise, chest pain, dyspnea, night sweats, and hemoptysis. Immunocompetent patients may have a self-limited disease that resolves over weeks to months, even without antifungal therapy.
• In the immunocompromised host, cryptococcal infection is often disseminated at the time of diagnosis, and patients most commonly present CNS findings, particularly meningoencephalitis, in addition to fever and cough. A very small minority of immunosuppressed patients presented with pulmonary symptoms, although severe cases may present with acute respiratory failure.25–28
Diagnostic Testing
• Definitive diagnosis of pulmonary cryptococcosis requires culture and identification of the organism from a sterile specimen but sensitivity may be poor.
• Cryptococcal antigen is found in the serum and CSF after the disease has become disseminated, and is of limited utility in immunocompetent patients with localized pulmonary disease. This test seems to be more sensitive in those with significant immunocompromise.25,28
• Cultures of BAL fluid increase the yield to almost 90%, and microscopic examination of the BAL smear with India ink may allow for rapid diagnosis. Testing of BAL fluid or sputum samples for cryptococcal antigen has yielded inconsistent results.26,28
• Radiographic findings of pulmonary cryptococcosis are variable, and dependent on the degree of immunosuppression in the patient.25,26,28
 In immunocompetent patients, solitary or multiple pulmonary nodules and airspace consolidation are the most common finding on CXR.
In immunocompetent patients, solitary or multiple pulmonary nodules and airspace consolidation are the most common finding on CXR.
 In immunosuppressed patients, diffuse, interstitial, and lobar infiltrates are the most common findings.
In immunosuppressed patients, diffuse, interstitial, and lobar infiltrates are the most common findings.
 Cavitation of pulmonary nodules and consolidations are more likely in patients with immunocompromise.
Cavitation of pulmonary nodules and consolidations are more likely in patients with immunocompromise.
Treatment
Because the risk of dissemination and recurrence is related to host immunity, treatment is based upon the status of patient immune function as well as the severity of disease. Treatment recommendations are outlined in Table 15-2.29
TABLE 15-2 TREATMENT RECOMMENDATIONS FOR PULMONARY CRYPTOCOCCOSIS

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


