Fig. 3.1
Formation of the heart from the first and second heart fields. Schematic representation of cardiogenesis showing the contribution of the first heart field (FHF – red), located in the cardiac crescent, to the myocardium of the left ventricle and other parts of the heart and the anterior second heart field (aSHF – brighter green) and posterior second heart field (pSHF – dark green) to the myocardium of the arterial and venous poles of the heart, as well as the right ventricle and atria. Abbreviations: LA left atrium, RA right atrium, LV left ventricle, RV right ventricle, OFT outflow tract, ao aorta, pt pulmonary trunk, PV pulmonary vein, RSV right superior caval vein, LSV left superior caval vein, E embryonic day of mouse development
Cardiomyocytes proliferate within the heart tube, and it was thought previously that this accounted for heart growth, with different regions of the early tube prefiguring the future atria and ventricles, in a segmental model of cardiogenesis [4]. However classic experiments in the chick embryo had already pointed to the addition of cells to the growing tube, notably to the inflow region [2]. Investigators subsequently showed that cells are added to the outflow region of the avian heart from adjacent mesoderm [5, 6]. At the same time, in 2001, addition of cells from pharyngeal mesoderm, marked by expression of a transgene integrated upstream of the fibroblast growth factor (Fgf) 10 locus, was demonstrated to contribute to the outflow tract and right ventricular myocardium in the mouse embryo [7]. The transcription factor Islet1 also marks these cells, and its expression pattern, together with the Islet1 mutant phenotype, indicated a contribution to the venous as well as the arterial pole of the heart [8]. This was confirmed later by explant experiments and tracking of cells labelled by dye injection in cultured embryos [9], which also showed that the early heart tube has a mainly left ventricular identity [10]. These findings regarding cardiac progenitor cells that form the developing heart tube were complemented by a different experimental approach examining myocardial cell lineages in the mouse embryo. Retrospective clonal analysis demonstrated that myocardium derives from two cell lineages. The first lineage is the source of left ventricular myocardium, whereas the second lineage is the source of the myocardium of the outflow tract and most of the right ventricle and also contributes to the atria which are derived from both lineages [11]. Thus, cardiac progenitor cells of the first heart field (FHF) form the early cardiac tube, whereas cells of the second heart field (SHF) subsequently enter the developing tube and constitute the second lineage contribution to the heart [2] (Fig. 3.2).
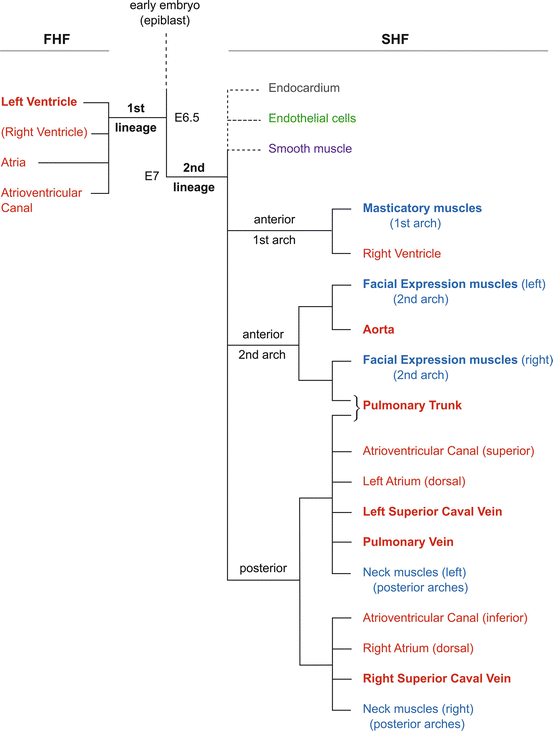
Fig. 3.2
A lineage tree for the heart. Schematic representation of the contribution of first and second cell lineages to the mouse heart. Myocardial derivatives are in red and skeletal muscle in blue, with bold type indicating that this is the major source, for example, the 1st lineage/first heart field for the left ventricle or the 2nd lineage/second heart field for head muscles, whereas not all neck muscles derive from this source. Other 2nd lineage (SHF) derivatives are indicted with dotted lines since their precise relation to each other or to the tree for myocardium and skeletal muscle is not yet clearly defined – endocardium in brown, endothelial cells in green and smooth muscle in purple. The approximate timing of lineage segregation from a common progenitor present in the epiblast is indicated. Abbreviations: FHF first heart field, SHF second heart field. E embryonic day of mouse development
The distinction between these two cell populations is underlined by the early segregation of the first and second lineages, previously estimated to take place at the onset of gastrulation or earlier [11]. This conclusion was drawn from a genetic approach to clonal analysis that depends on a random recombination event in a progenitor cell that converts an inactive laacZ reporter to an active lacZ sequence. In this case the reporter was targeted to an allele of the cardiac actin gene resulting in b-galactosidase expression in clonally related daughter cells in the myocardium. The size of the clone gives an estimation of the time of recombination. However this is only approximative. Recently [12], another genetic approach to clonal analysis made it possible to have a more precise estimation of the timing of clonal segregation and also to follow all mesodermal cell types in the heart. This approach is based on a conditional confetti multicolour reporter and brief induction of a Cre recombinase that activates a single coloured reporter in a progenitor cell and its descendants. In this case the Cre is controlled by the mesoderm posterior 1 homolog (Mesp1) gene which is expressed in the primitive streak in gastrulating mesodermal cells that will contribute to all compartments of the heart [13]. These experiments confirmed that two myocardial cell lineages form the heart, as previously described, and demonstrated that the first lineage can be distinguished by E6.25–E6.75, at the onset of gastrulation, whereas the second lineage that gives rise to SHF derivatives is marked when the Cre is induced at E6.75–E7.25. A similar genetic approach based on a mosaic analysis with double markers (MADM) also showed distinct segregation of two lineages [14]. The common progenitor that gives rise to the two lineages is therefore present prior to activation of Mesp1 early in gastrulation, before day 12 of human embryonic development.
3.2 The First Heart Field
The first differentiated cells expressing cardiac muscle markers are detected prior to formation of the heart tube, in a region under the head folds that spans the midline of the embryo, known as the cardiac crescent. The early heart tube forms as a result of fusion of the crescent at the midline. This process reflects morphogenetic movements of the underlying endoderm as the embryo develops but also depends on genes expressed in cardiac mesoderm which control cell migration and fusion of the crescent. Not much is known about the underlying cell biology in mammals; however a number of mutations have been identified that perturb the process. Failure to migrate leads to cardiac bifida where two hearts begin to develop on either side of the embryo. Mutation of the gene encoding the transcription factor GATA binding protein 4 (Gata4) provides a classic example of this phenotype [2]. The early heart tube expresses transgenic markers for left ventricular myocardium, and dye labelling of these cells followed by embryo culture showed that they will mainly contribute to the left ventricle [10]. Markers of cells in the crescent that distinguish the first from the second heart field have been elusive. For example, the transcription factor T-box (Tbx) 5, implicated in Holt-Oram syndrome [15], is present in FHF cells, but it also marks subdomains of the SHF [16] and is expressed dynamically in the forming heart tube before showing left ventricular localisation. The hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 (Hcn4) gene which is expressed later in the conduction system is transitorily activated specifically in the cardiac crescent and thus provides an interesting early marker [17]. However it is probably expressed in myocardial cells at this stage rather than in the FHF progenitors. Induction of a Mesp1–GFP (green fluorescent protein) transgene at the early (E6.5) and later (E.7.5) time points, when progenitors for the first and then the second heart fields can be distinguished [12], makes it possible to separate these cell populations by flow cytometry and to perform transcriptome analysis. This approach has already identified a number of differentially expressed genes and should lead to novel markers. Such experiments also open the way to functional analysis, not only for FHF cells present in the crescent but also at earlier stages as they progress through the primitive streak, exit it and then migrate anteriorly to take up their position under the head folds.
3.3 The Second Heart Field
The cardiac progenitor cells of the SHF (Fig. 3.1) are located medially to the cardiac crescent, extending posteriorly. As the crescent fuses to form the early heart tube, these cells come to lie behind the tube and also anteriorly, adjacent to the outflow tract, as well as posteriorly, adjacent to the sinus venosus and venous pole as it develops. Initially the tube is not completely closed, and there is continuity with the underlying dorsal mesocardium which is attached to the pericardial wall, with some differentiating cells entering the heart dorsally from the SHF [11]. Subsequently the dorsal mesocardium breaks down, as the tube closes. The cardiac tube becomes suspended in the pericardial cavity, with cells of the SHF continuing to feed into the poles of the heart to form myocardium. This process continues over several days in the mouse embryo, with myocardium at the base of the pulmonary and caval veins still being formed as late as E12.5 (40 days of human development). The SHF provides a pool of cardiac progenitor cells which are maintained as a proliferating, non-differentiated population. Transcription factors and signalling pathways that mark these cells [18] play a critical role in regulating this behaviour. Mutations in these genes lead to major arterial pole defects and also to venous pole defects which may be more subtle, reflecting the fact that the outflow tract is derived exclusively from the second myocardial cell lineage and thus is dependent entirely on the SHF contribution. In newborn children with a congenital heart defect, 30 % have a malformation of the arterial pole of the heart. Fortunately this can usually be corrected surgically and the child survives. However, a small proportion of babies die after surgery, and re-examination of the records indicates that in a number of cases there was also a venous pole defect that had not been detected [19]. It is therefore very important to check for subtle venous pole defects which can be corrected at the same time as surgery is performed to correct a defect at the arterial pole.
3.3.1 Regulation of Proliferation and Differentiation
Cells in the SHF are highly proliferative so that the pool of cardiac progenitor cells is maintained as the heart grows. FGF, canonical WNT and Hedgehog signalling promote proliferation [18]. Differentiation of SHF cells is triggered as they enter the poles of the heart. BMP signalling plays an important role in this process, antagonising FGF signalling [20, 21] and promoting the deployment and differentiation of cells that will contribute to the arterial pole. Notch and non-canonical WNT signalling are implicated also in cardiac differentiation [18]. At the venous pole of the heart, canonical WNT, Hedgehog and BMP signalling orchestrate the critical balance between proliferation and differentiation, where Hedgehog signalling promotes the migration of cells to the heart tube as well as their proliferation [22, 23]. These pathways are activated by signals from surrounding tissues as well as by cell autonomous signalling within the SHF [24]. Thus, the midline structures (neural tube and notochord) are sources of canonical WNT and Hedgehog signalling, promoting SHF proliferation. Hedgehog signalling also comes from the endoderm, while FGF signals come from surrounding endoderm and ectoderm as well as from the SHF itself. Neural crest cells, which migrate through the SHF into the arterial pole, mediate BMP inhibition of FGF signalling, such that in the absence of neural crest, excessive SHF cell proliferation results in reduced cardiomyocyte differentiation and defective growth of the heart. BMP signals are also produced by cells in the SHF, promoting neural crest survival, and by cardiomyocytes, promoting recruitment to the poles of the heart.
An extensive gene regulatory network operates in the SHF to control cardiac progenitor cell behaviour [18]. Key transcription factors modulate signalling pathways, illustrated by Tbx1 interference with the BMP signalling cascade to delay differentiation [24, 25] or Tbx1 activation of enhancers that control Fgf8 and Fgf10 expression in the SHF [24–26]. Islet1 also activates the Fgf10 enhancer [26] and has a SHF enhancer which is itself controlled by forkhead box (Fox) c2 [18].
Some genes expressed in the SHF are specific to the progenitor cell population in this cardiac context. Foxc2, for example, marks SHF progenitors, and Islet1 has long been considered to be the key marker of this field. However, more sensitive reporters now indicate that Islet1 is also expressed at a low level in FHF progenitors, although the Islet1 mutant phenotype suggests that it is mainly functional in the SHF [18].
Genes that are involved in myocardial differentiation also are expressed in the SHF. These include genes coding for the transcription factors, NK2 homeobox 5 (Nkx2-5), Gata4, Mef2c, serum response factor (SRF), Tbx5 and the chromatin remodelling protein SWI-/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily D, member 3 (Smarcd3; also called 60 kDa BRG-1/Brm associated factor subunit C1; Baf60c) which marks a subset of early Mesp1-positive cardiac progenitors [18]. These genes are expressed in differentiating cells of the cardiac crescent and in regions of the heart, where combinations of them activate sarcomeric genes. Ectopic expression of Gata4, Tbx5 and Smarcd3 in non-cardiogenic regions of the mouse embryo has been shown to activate cardiac muscle formation [27], indicating the role of these factors in cardiac progenitor cell determination as well as in cardiac differentiation. Their targets in the SHF are not well understood, although transcriptome analysis of Nkx2–5-expressing cells gives some insights [28]. Notably, Nkx2–5 functions in a negative feedback loop with Bmp2/Smad1 signalling that controls cardiac progenitor cell behaviour, which is probably of importance in the aetiology of Nkx2-5-related human congenital heart defects. Different combinations of factors are one reason why targets may differ between cardiac progenitors and myocardium. Another consideration is the level of expression which may affect function. The potential importance of modulating the level of these factors is indicated by Tbx1 downregulation of SRF and Tbx5 in the SHF [16, 25]. In the Tbx1 mutant, abnormal cardiac differentiation of SHF progenitors in the pericardial wall probably reflects this effect as well as deregulation of the balance between BMP and FGF signalling [24]. The SHF enhancer of the Fgf10 gene provides an example of how levels and combinations of transcription factors can be functionally important [26]. In this enhancer both Islet1 and Nkx2-5 bind to the same sites. In the SHF, Islet1 levels are high, and it competes successfully for binding, leading to activation of Fgf10, which also depends on Tbx1. In the heart, on the other hand, Nkx2-5 levels are very high and it displaces Islet1. This leads to downregulation of Fgf10 in keeping with the repressive function of Nkx2-5 observed for SHF genes, including Islet1, in the heart [28]. Since Nkx2-5 also functions as a transcriptional activator in the myocardium this dual role again depends on combinatorial effects of other factors.
Most transcription factors and components of signalling pathways that play a role in cardiogenesis are not cardiac specific. This is in contrast to skeletal muscle where the myogenic regulatory factors of the myogenic differentiation (MyoD) family are only expressed during myogenesis [29]. Analysis of mutant phenotypes is informative for the heart only when the gene does not play a vital role during early embryogenesis. Islet1 or Mesp1, for example, is mainly important for formation of the heart at early stages of development, whereas Fgf8 is required for gastrulation and conditional mutation of Fgf8 directed to cardiac progenitors is essential in order to understand its role in the SHF. Mesp1-Cre lines [13] have proved to be very valuable in targeting all cardiac progenitors, whereas an enhancer of the Mef2c gene targets cells in the SHF [30]. It follows that in the human population, many mutations in cardiac regulatory genes may be early embryonic lethal or result in multiple effects as seen in a number of syndromes where the heart is affected. This is the case, for example, of mutations in TBX1 which give rise to DiGeorge syndrome [31]. This includes major defects at the arterial pole of the heart, such as common arterial trunk or persistent truncus arteriosus as well as craniofacial abnormalities, due to the function of Tbx1 in the SHF as well as in endoderm and ectoderm in the pharyngeal region which is important for cardiac and cranial neural crest. Unless there is some cardiac-specific aspect of function, mutations in enhancers that direct gene transcription to the cardiac progenitors of the first or second heart fields are the best candidates for defects that are restricted to the formation of the heart. Many such enhancers, that specifically direct transcription in the SHF, have now been identified for genes such as Fgf8, Fgf10, Islet1, Nkx2–5 or Mef2c [18]. They are regulated by combinations of SHF transcription factors, as for Fgf10 [26].
3.3.2 Patterning the SHF
The SHF is patterned on the anterior/posterior axis. Although gene expression has not yet been compared at a single-cell level, regional heterogeneity is apparent. The anterior part of the SHF is marked by genes that are not expressed throughout the field [18]. This is the case for Fgf8 or Fgf10 and also for the SHF Mef2c enhancer which is not active in the posterior SHF. This anterior/posterior distinction correlates with the contribution of cells expressing these genes to the arterial or venous pole, respectively. This is illustrated in the anterior SHF also, where Tbx1 is mainly expressed in cells that will contribute to myocardium at the base of the pulmonary trunk [24]; in contrast to Fgf8/10 or the Mef2c enhancer which mark progenitors of the right ventricle as well as outflow tract myocardium, both of the pulmonary trunk and the aorta. Differences in the origin of myocardium at the base of these arteries are reflected by differences in gene expression [16]. Pulmonary trunk myocardium, for example, is marked by semaphorin 3c (Sema3c) gene expression. In Tbx1mutants pulmonary trunk myocardium is reduced [24], which is probably the early abnormality that underlies the development of tetralogy of Fallot and other cardiac defects in DiGeorge syndrome patients.
The posterior SHF is marked also by distinct gene expression profiles. Homeobox (Hox) genes, differentially expressed along the axis of the embryo, are classically involved in anterior/ posterior patterning. Anterior members of the Hox gene clusters such as Hoxb1 are expressed in the posterior SHF, under the control of retinoic acid signalling. When this signalling pathway is perturbed, Hox gene expression is affected, and the posterior boundary of the SHF is modified [35]. Tbx5 is expressed in cardiac progenitor cells in the posterior SHF. In addition to atrial myocardium, the dorsal mesenchymal protrusion [33] also derives from the posterior SHF, from cells expressing Tbx5 and Islet1, and its development depends on Hedgehog and BMP signalling [22]. This structure is important in the context of congenital heart malformations since it provides the muscular base of the atrial septum, and anomalies in its formation from the posterior SHF lead to atrial and atrioventricular septal defects. Tbx18 is expressed in a particular lateral domain of the posterior SHF which is distinguished also by lack of Nkx2–5 and Islet1 expression [34]. This domain, which has been proposed to constitute a third heart field, contributes caval vein myocardium, which is marked by continuing Tbx18 expression. Retrospective clonal analysis distinguishes sub-lineages of the second myocardial cell lineage (equated with the SHF) which contribute to myocardium at the arterial or venous poles of the heart. However this clonal analysis does not distinguish a particular sub-lineage contributing to caval vein myocardium only, as might be expected if this arises from a third heart field [35].
The distinction between anterior and posterior domains of the SHF is made on the basis of gene expression; however, cells may move between these domains. This was first indicated by genetic tracing of cells that had expressed Hoxb1 in the posterior SHF [32], leading to labelling of myocardial derivatives at the venous pole. Unexpectedly Tbx1-dependent myocardium at the base of the pulmonary trunk was labelled also [16]. In keeping with this result, dye labelling of small groups of cells in the posterior domain of the SHF showed, after embryo culture, that some labelled cells were located in the outflow tract and had therefore moved anteriorly. In so doing these cells activated markers of the anterior SHF, as indicated by Fgf10 reporter gene expression [36]. Most cells do not appear to move extensively within the SHF and clonal analysis [2] suggests coherent, rather than dispersed, cell growth as the heart forms. However lineage studies for the venous pole of the heart showed unexpected clonality between myocardium at the left side of the venous pole (pulmonary vein, left caval vein and left atrium) and pulmonary trunk myocardium at the arterial pole [35]. This is consistent with anterior movement of a subset of progenitor cells located in the left side of the posterior SHF from which left venous pole myocardium derives. Unexpectedly, Cre genetic tracing experiments show that the classic anterior SHF markers, Tbx1 [37] and the Mef2c–AHF enhancer [16], are transiently expressed in progenitor cells giving rise to the atrial septum and dorsal mesenchymal protrusion. Indeed, recent experiments have shown that loss of function of Tbx1 impacts not only the anterior SHF but also the posterior SHF, resulting in venous as well as arterial pole defects [16]. The development of the dorsal mesenchymal protrusion is affected, leading to ostium primum and atrioventricular septal defects. Some anterior SHF cells may move posteriorly to contribute to parts of the venous pole; however, there is no evidence to date for this. Alternatively, diminished proliferation and premature differentiation of cells in the SHF in the absence of Tbx1 may perturb the demarcation between anterior and posterior regions of the SHF. In addition Tbx1 may play a role in the segregation of cells to anterior and posterior domains from a common early progenitor population.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


