Fig. 8.1
Estimated prevalence of elevated Pi, PTH and FGF23 across the strata of chronic kidney disease. FGF23 is an early biomarker of renal dysfunction, and its increase precedes the development of secondary hyperparathyroidism. Hyperphosphatemia is usually prevented by these homeostatic mechanisms until CKD stage 4 or 5 (GFR < 30 ml/min/1.73 m2). Adapted from the CRIC cohort
In addition to stimulation by Pi, calcium and PTH, a decline in Klotho, a membrane-bound co-receptor for FGF23, is likely contributing to excessive FGF23 production in advanced CKD due to end-organ (i.e. renal) FGF23 resistance. This is analogous to high insulin concentrations observed in type 2 diabetic patients to overcome peripheral insulin resistance. The residual expression of Klotho is markedly reduced in CKD with less than 10 % of normal levels in many ESRD subjects [10]. In the context of mineral metabolism, this accelerates and aggravates the typical biochemical profile of high FGF23, sHPT, low 1,25(OH)2D and hyperphosphatemia.
A key question is whether these biochemical abnormalities should be considered merely as cosmetic defects, or if they actively contribute to mechanisms of CVD. A wealth of clinical and epidemiological data supports the latter. Even mild renal dysfunction is independently associated with increased CVD risk, and cardiovascular mortality is 10–30 times higher in ESRD patients on dialysis treatment compared to the general population [11]. A more descriptive and complete discussion of cardiovascular pathology in relation to CKD are provided elsewhere in this book, yet we herein want to emphasize two dominant CVD features in the context of serum Pi and FGF23: vascular calcification with stiffening of the arteries and left ventricular hypertrophy/congestive heart failure.
Hyperphosphatemia and Cardiovascular Risk
Over the last decades a link between serum Pi and cardiovascular risk has gradually been uncovered. This relationship was noted early by Block and colleagues in ESRD patients, which is the most common clinical situation of severe hyperphosphatemia [12]. With adjustment for case mix they found a graded relationship between increasing serum Pi levels and mortality risk and cardiovascular hospitalization, which remained significant after multivariate adjustment for other confounding CVD risk factors. Another hallmark study conducted by Tonelli and co-workers demonstrated a similar graded relationship between serum Pi levels and cardiovascular risk in individuals free of CKD [13]. It is noteworthy that this association held true in a setting of Pi levels within the defined normal reference range. Similar findings have subsequently been corroborated in various CKD studies, in renal transplant recipients and in community-based cohorts as well as in the general population [14–17].
Due to the complex interplay and dynamics of mineral metabolism in relation to CKD, and the large risk for residual confounding in epidemiological settings, some concerns about true causality have been voiced. Yet, there is an overall consensus that serum Pi is a considerable risk factor in CKD, not at least due to the consistency of data pointing to serum Pi as an independent cardiovascular risk factor, and because the association between hyperphosphatemia and mortality appears stronger than for other components of mineral metabolism. Indeed, a recent meta-analysis comprising more than 300,000 CKD patients demonstrated that a 1 mg/dL increase in serum Pi was associated with an 18 % higher risk of death, whereas serum calcium and PTH were not associated to increased mortality risk [18].
What is then the mechanisms linking hyperphosphatemia to CVD and mortality? Several biologically plausible, and experimentally proven, pathways by which Pi promotes CVD have been proposed [19, 20]. First, under conditions of hyperphosphatemia the rate of Pi influx from extracellular fluids into various cell types, including vascular smooth muscle cells (VSMC), are augmented. This is accomplished by an active, sodium-dependent Pi transport of the ubiquitously expressed Pit-1 protein. Downstream consequences of augmented Pi uptake in VSMCs include cell death and osteochondrocytic differentiation. Osteochondrocytic differentiation is a maladaptive process where VSMCs transform to an osteoblast-like phenotype, associated with local production of bone proteins and release of precalcified membrane matrix vesicles. Under normal circumstances these vesicles contain mineralization inhibitors such as Fetuin A and matrix Gla protein, and are believed to protect the VSMC from calcium overload. In CKD these inhibitors are low or absent, thus turning the vesicles into foci for calcification. Vascular calcification is indeed a dominant feature in a majority of CKD patients [21], and the presence and severity of vascular calcification increases in parallel with serum Pi (Fig. 8.2) [22, 23]. Notably, the ability of Pi to induce vascular calcification diminishes drastically in the absence of calcium [24]. Inhibition of the formation of Ca-Pi crystals effectively prevent the progression of Pi-driven vascular calcification as well [25], supporting that Pi and calcium operate synergistically in the calcification process. This concept is also supported by several randomized clinical trials (RCTs) that typically demonstrate a relative decline in the progression rate of vascular calcification when using non-calcium based Pi binders compared to calcium-based treatment regimens [26, 27].
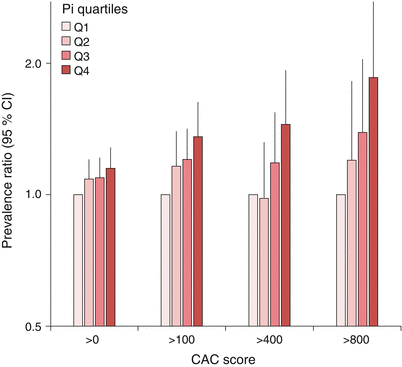
Fig. 8.2
Higher serum Pi is associated with the adjusted prevalence of increased coronary artery calcification (CAC) score. Adapted from the CRIC study.
It should be underscored that high serum Pi may have other detrimental consequences beyond vascular calcification. Rapid infusions of Pi in rodents and humans have shown an acute impairment in the endothelial response to other vasoactive substances, and epidemiological studies in CKD patients confirm an independent association between serum Pi and endothelial dysfunction, the latter being a well-established surrogate marker of long-term cardiovascular risk [28–30]. Similarly, hyperphosphatemia is associated with both static and dynamic measurements of cardiovascular function including impaired endothelium-dependent and endothelium-independent vasodilation, increased pulse-wave velocity (a measure of vascular stiffness), carotid artery intima-media thickness, as well as measurements of systolic and diastolic cardiac dysfunction [13, 16, 31, 32].
In experimental models, Pi loading has many detrimental consequences, both at the cellular and organ level. First, high Pi induces cellular senescence defined by telomere shortening and apoptosis [33]. Second, it induces fibrotic processes through several different pathways, and phenotypic abnormalities promoted by Pi can be ameliorated or reversed by dietary restriction of Pi and/or Pi binding therapy, strengthening the likelihood of causal relationships in epidemiological studies [34, 35]. Fibrosis is the end result of a chronically failing nephron, and a pro-fibrotic environment accelerates the underlying progression rate in CKD. It is intriguing that recent clinical data not only confirm a link between serum Pi and CVD but support an association between serum Pi and an accelerated CKD progression rate [36, 37]. Another organ-specific consequence of pro-fibrotic stimuli is cardiac fibrosis, frequently occurring in conjunction with development of left ventricular hypertrophy (LVH). LVH is a common complication in CKD patients and is a strong predictor of cardiac events and mortality in the general population as well as in CKD patients [38, 39].
Despite cumulative evidence favoring serum Pi as a cardiovascular toxin, few RCTs have properly examined whether Pi-lowering therapy improves long-term cardiovascular outcome. The largest RCT to date that addressed this hypothesis was conducted in dialysis patients and did not show any survival benefit in the primary analysis when comparing treatment with sevelamer (a non-calcium Pi binder) and calcium-based Pi binders [40]. Interpretation of data is hampered by several factors attributed to design and execution of this study, not at least the remarkable drop-out rate of approximately half of all subjects randomized to treatment. Subsequent head-to-head RCTs comparing sevelamer with Ca-based Pi binders have convincingly demonstrated a reduction in the progression rate of vascular calcification as measured by abdominal calcification score (ACS) or coronary artery calcification score (CAC) [26, 41]. Although less calcification is expected to translate into reduced long-term cardiovascular risk, this remains an open issue. A few small studies comparing sevelamer and Ca-based Pi binders supported a survival benefit for sevelamer, although these studies were not adequately powered to assess the impact on mortality [42, 43]. A recent meta-analysis including more than 4600 patients from 11 different trials indicated a reduction in all-cause mortality by 22 % in favor of non-Ca Pi binding treatments [44]. As a note of caution, none of these studies were placebo-controlled, implying that the true advantage of Pi-lowering therapy remains unclear. A possibility is that the assumed survival benefit attributed to non-Ca Pi binders could be related to less Ca exposure rather than a reduction in intestinal Pi absorption. In this regard, a recent placebo-controlled RCT which explored the efficacy of various Pi binders for slowing progression of vascular calcification in non-dialysis normophosphatemic CKD patients demonstrated no benefit, but rather a trend towards harm, for Pi binders as compared to placebo [45]. Although this trend appeared primarily driven by Ca-based Pi binders, it is still possible that gastrointestinal Ca absorption (and thus systemic Ca exposure) is augmented by Pi binding therapies in this setting. The reason is because the intestinal fraction of free Ca increases when Pi is attached to the binder rather than forming fecally excreted Ca-Pi complexes. Nevertheless, this study underpins the need for more rigorous exploration of serum Pi as a modifiable cardiovascular risk factor.
FGF23 and Cardiovascular Risk
Considering the dynamics of serum FGF23 and Pi in CKD depicted in Fig. 8.1 (e.g. FGF23 rises in early CKD whereas Pi levels are maintained normal until advanced CKD or ESRD due to homeostatic mechanisms) the idea was planted that FGF23 may be a surrogate marker for time-averaged Pi exposure, analogous to Hb1Ac and glucose levels. This concept was fueled in a study from 2008 by Gutierrez et al. that provided the first clinical evidence for FGF23 as an independent predictor of mortality in incident hemodialysis patients [46]. The magnitude of this relationship was strong, with a nearly six-fold increased risk for mortality during the first year of hemodialysis in those individuals within the highest quartile of FGF23 compared to those in the lowest. Equally important, a relationship between FGF23 and mortality was confirmed irrespective of baseline serum Pi levels, and the predictive value of FGF23 was superior relative to serum Pi. This report was followed by a multitude of observational studies confirming FGF23 as a marker of mortality across the spectrum of CKD, including early and late stage CKD, incident and prevalent dialysis populations and renal transplant recipients [47–49]. Similarly as for serum Pi, several reports confirmed that FGF23 predicted mortality in community-based settings and in individuals free of CKD [50, 51].
Epidemiological studies do not prove causality, but they may shed light on potentially relevant mechanisms that drive observational findings. Subsequent data more specifically supports a link between FGF23 and CVD, and that FGF23 is more robustly linked to cardiovascular mortality than non-cardiovascular or total mortality. In an attempt to dissect which aspects of cardiovascular pathology FGF23 portrays, we initially measured FGF23 in the PIVUS study, which is a community-based, prospective, observational study in which all study participants have undergone careful evaluation of subclinical indices of cardiovascular function. In this study population, FGF23 was associated with impaired dynamic vasoreactivity and endothelial function, total atherosclerosis score, LVMI and risk for the presence of LVH [52–54]. All these associations remained significant after adjustment for relevant cardiovascular risk factors and for markers of mineral metabolism including serum Pi. Subsequent studies in various CKD populations have confirmed these findings and also demonstrated that FGF23 predicts new onset or aggravation of pre-existing conditions in longitudinal follow-up studies, in particular CHF and LVMI/LVH [55–58]. In the PIVUS study a 10 % increase in FGF23 corresponded to a 0.7 % increase in LVMI. Notably, this strength of relationship was observed in a setting of essentially normal renal function (mean eGFR 77 ml/min/1.73 m2). In comparison, in patients from the CRIC study (average eGFR 42 ml/min/1.73 m2) every unit increase in natural log-transformed FGF23 associated with a 5 g/m2.7 increase in LVMI, translating to approximately 0.9 % increase in LVMI per 10 % increase in FGF23. In principle, this means that a reduction in FGF23 by 20–40 % that may be accomplished by cinacalcet or phosphate-binding treatment in some CKD patients potentially could result in a clinically significant reduction of LVMI, although not of the same magnitude as for anti-hypertensives (−6–13 %) [59]. In contrast to serum Pi, FGF23 is not consistently linked to vascular calcification. Recent data from the CRIC study showed that FGF23 were not associated with coronary artery or thoracic aorta calcium in a cohort of 1,501 CKD patients [23].
In principle, the current situation must be dealt with: serum Pi and FGF23 are both independent predictors of hard cardiovascular endpoints as well as intermediate/surrogate cardiovascular endpoints. There is no critical effect modification by any of the parameters, implying that both factors portray risk independent of the other. This leaves us with two questions, one being if FGF23 only is a biomarker of CVD? If the answer is no, is then serum Pi and FGF23 two cardiovascular toxins with similar or distinct toxicity profiles?
Is FGF23 a Biomarker or Contributor to CVD?
Based on elegant experimental work, it has been established that membrane-bound alpha-Klotho (Klotho) is a permissive co-receptor for FGF23 that allows its high-affinity binding to cognate FGF-receptors (FGFRs) [60]. According to the initial Klotho reports originating from 1997 onwards, its expression is largely confined to renal tubules, parathyroid glands and choroid plexus [61], but not within the cardiovascular system except the sinoatrial node of the heart [62]. Given the absence of its co-receptor in the cardiovascular system, the prevailing assumption has been that FGF23 cannot directly contribute to CVD. However, this concept has recently been challenged by studies providing evidence for Klotho expression in the vasculature and the presence of alternative (Klotho-independent) FGF23 signaling pathways in the heart.
Lim et al. report that Klotho is expressed in human VSMCs, and that FGF23 attenuate vascular calcification in vitro [63]. Further, they show that vascular Klotho declines in parallel with renal function, which would explain the loss of FGF23’s anticalcific effects in CKD. This report was later substantiated by other studies reporting on vascular Klotho expression in rodents and humans [64, 65]. Second, Faul et al. demonstrate that FGF23 induces LVH in mice, and that treatment with FGF23 directly stimulates growth of isolated cardiomyocytes in vitro [56]. Klotho is not expressed in cardiomyocytes and the effects of FGF23 on the heart were shown to be mediated via the non-canonical PLCγ-calcineurin pathway. Moreover, the effects of FGF23 on LVH were blocked by administration of an FGF-receptor inhibitor.
However, both these mechanisms remain controversial and subsequent studies have failed to replicate the findings. As previously mentioned, Scialla et al. did not see Klotho expression in human or mice VSMCs or in mouse aorta, nor any direct effects of FGF23 on the calcification process in vitro [23]. Similar data were also reported in a study by Lindberg et al., in which Klotho expression was very low or absent in major murine arteries, and FGF23 lacked effects on endothelial function and vascular calcification [66]. On the other hand, if FGF23 can signal via non-canonical pathways in the heart, cannot that also be the case in the vasculature? In the context of vascular effects of FGF23, the presence or absence of Klotho may perhaps be more of an academic discussion if FGF23 targets the vasculature through alternate signaling mechanisms. Regardless of which pathway FGF23 acts on, data on direct effects on the vasculature is scarce and conflicting, and further studies are warranted.
Concerning FGF23 and cardiomyopathy, a recent study by Agerwal et al. failed to confirm an association between elevated Fgf23, LVH and reduced ejection fraction in Klotho knockout mice, and only found a weak relationship between FGF23 and left ventricular ejection fraction in patients free of CKD [67]. Similarly, in a study by Shalhoub et al. treatment of uremic rats with neutralizing FGF23 antibodies had no effects on LVH or cardiac hypertrophy markers [8]. Another dilemma is that patients with certain endocrine disorders characterized by over-expression of FGF23, such as tumor-induced osteomalacia and X-linked hypophosphatemic rickets, do not suffer from overt LVH or higher risk for CVD. That does not however fully exclude the possibility that FGF23 is harmful, since its absolute levels are much higher in ESRD subjects and its detrimental effect may be potentiated in the uremic environment.
As a final remark on this topic, it should be noted that the current understanding of FGF23 in relation to CVD may be confounded by its co-receptor Klotho. Some studies have shown that soluble Klotho (a shedded form of membrane-bound Klotho) inhibits vascular calcification and progressive renal damage, at least partly through inhibition of TGF-β and Wnt signaling [68–70]. The inverse relationship between FGF23 and membrane-bound Klotho in CKD implies that Klotho deficiency, rather than FGF23 excess, may explain some of the aforementioned epidemiological findings. Unfortunately, methodological limitations in measuring soluble Klotho hamper clinical research in this field at present.
An Emerging Model of Serum Pi and FGF23 as Two Distinct Cardiovascular Toxins
Consolidating the rich flora of clinical and experimental data, we propose a model in which most definitely serum Pi, and quite likely FGF23, are to be considered cardiovascular toxins. Their toxicity profiles may overlap, yet a pattern can be discerned in which serum Pi at first hand promotes vascular calcification whereas FGF23 may drive LVH. This hypothesis is summarized in Fig. 8.3. The most severe clinical consequences of LVH are CHF and risk for arrhythmias and sudden death. In this context, a recent preliminary post-hoc analysis of the EVOLVE trial suggesting that individuals with a large reduction in serum FGF23 upon treatment with the calcimimetic cinacalcet had a markedly reduced event rate for CHF is of prime interest [71].
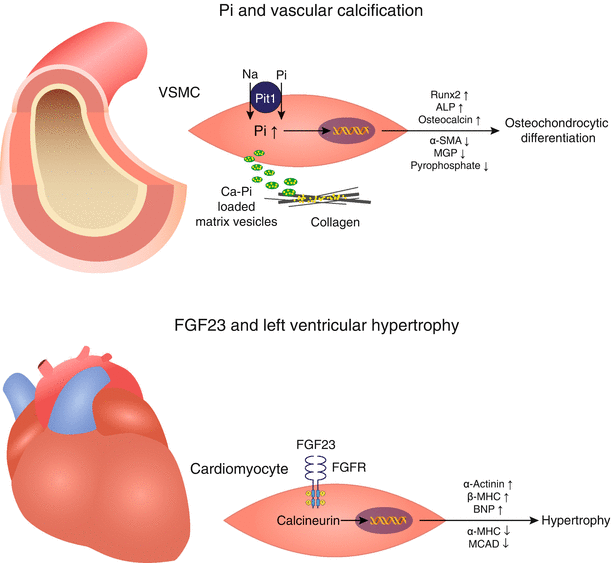
Fig. 8.3
Proposed model: FGF23 and phosphate – two cardiovascular toxins with distinct toxicity profiles. Top: A wealth of epidemiological and experimental data has implicated a direct role for Pi in cardiovascular pathology, particularly in the development of vascular calcification. Intracellular excess of Pi in vascular smooth muscle cells induces osteochondrocytic differentiation and release of calcified matrix vesicles. Bottom: Novel data suggest Klotho-independent effects of FGF23 on cardiomyocytes, resulting in cell growth and development of left ventricular hypertrophy
Summary and Conclusion
Epidemiological and RCT data consistently link hyperphosphatemia and high FGF23 to increased mortality risk and numerous long-term or surrogate cardiovascular endpoints. The link between hyperphosphatemia and vascular calcification seems straightforward based on the collective record of experimental and clinical data as well as biological plausibility. The acceptance of serum Pi as a surrogate for vascular calcification and cardiovascular outcomes is evidenced by the widespread use of Pi binders in the CKD population, especially in dialysis patients [72]. Nevertheless, further RCTs are warranted to fully delineate the benefit of lowering serum Pi for reducing CVD, particularly in the non-dialysis CKD population.
The link between FGF23 and CVD is largely of epidemiological nature and unravel FGF23 as a relatively stronger predictor of CVD than serum Pi across all ranges of kidney function, perhaps with a more distinct cardiac toxicity profile. The clinical use of FGF23 as a biomarker for identification of high risk individuals and enrichment strategies in RCTs deserves further exploration. Identification of Klotho-independent FGF23 signaling in the heart is illuminating and presents novel plausible explanations for causal relationships between FGF23 and LVH/CHF, yet the relevance of FGF23 “off-target” signaling remains unclear and future research efforts should explore the presence, magnitude and relevance of FGF23 signaling in the cardiovascular system.
References
1.
2.
Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–8.PubMedCentralPubMedCrossRef
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


