Exercise and Rehabilitation in Congestive Heart Failure
Donna M. Mancini
Rebecca P. Streeter
The clinical syndrome of congestive heart failure (CHF) results in changes in the skeletal muscles, peripheral vasculature, and lungs that are a consequence of a chronically decreased cardiac output. Exercise training may be an effective therapeutic modality to retard or reverse the maladaptive changes that develop in the periphery during CHF (1). In this review, we summarize the central and peripheral adaptations encountered in CHF and discuss the prognostic use of exercise testing and the therapeutic potential of exercise training in patients with heart failure.
Cardiac and Peripheral Derangements in Heart Failure
In the presence of a reduced cardiac output, the heart depends on three principal compensatory mechanisms to maintain normal function (2). First, the Frank-Starling mechanism increases preload to sustain cardiac stroke volume. Second, myocardial hypertrophy develops to increase the mass of contractile tissue. Third, the sympathetic nervous system is activated to augment myocardial contractility. In the short term, these compensatory mechanisms serve to preserve cardiac output. However, eventually these compensatory mechanisms become detrimental, contributing to the progression of the disease process.
In normal subjects, the primary limitation to maximal physical performance is the cardiac-output response (2,3). Patients with heart failure exhibit reduced cardiac-output responses to exercise in comparison with normal subjects. The decreased exercise capacity of patients with heart failure has been attributed to a decreased cardiac-output response, which leads to inadequate perfusion of skeletal muscle and intramuscular lactic acidosis (4,5). In 1981, Weber et al. (6) measured the hemodynamic and ventilatory responses of 40 patients with heart failure during progressive treadmill exercise. This first large application of the measurement of peak oxygen consumption (VO2) in patients with heart failure demonstrated the usefulness of this technique as a noninvasive method for characterizing cardiac reserve and functional status. Weber et al. demonstrated a significant correlation between cardiac-output response and VO2 (Fig. 38-1) and were able to classify patients into groups with disease of increasing severity on the basis of this noninvasive technique.
Although peak exercise capacity clearly depends on the cardiac-output response to exercise, this is not the sole determinant of exercise performance in patients with heart failure. The exercise capacity of patients with severely reduced ejection fractions varies widely. Furthermore, therapeutic interventions aimed at acutely increasing cardiac output, such as administration of dobutamine, do not significantly increase peak VO2 (7,8). This discrepancy between enhanced cardiac output and fixed peak VO2 can be explained on the basis of the peripheral, vascular, and skeletal muscle derangements in CHF. Because of regional vascular or skeletal abnormalities, the augmented cardiac output cannot be utilized by the exercising muscle beds and, therefore, peak VO2 is not altered.
Peripheral Vascular Changes in Heart Failure
Experimental evidence suggests that the peripheral circulation undergoes substantial transformations during the progression of CHF, with an alteration of regional vascular control. The alterations occur both at the level of the vascular endothelium (an important modulator of vascular tone) and at the level of the vascular smooth muscle.
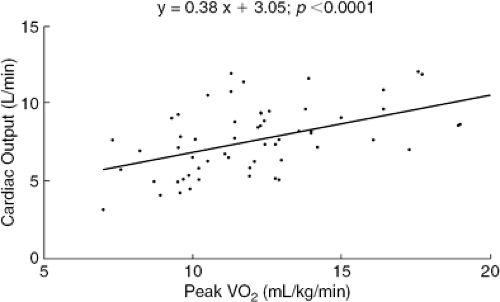 Figure 38-1 Correlation of peak exercise cardiac output with peak VO2. |
In both animal and human models of CHF, the responses to endothelium-mediated vasodilation are blunted. Endothelium-dependent dilation is reduced in the aorta of rats with ischemic cardiomyopathy (9). Kubo et al. (10) demonstrated that endothelium-dependent vasodilation is attenuated in patients with heart failure. Changes in sheer stress are an important determinant of endothelial function. In normal vasculature, the changes in sheer stress on the endothelial cells that accompany alterations in blood flow serve to enhance vasodilation via release of nitric oxide and prostaglandins (11,12). Endothelial dysfunction appears to be a time-dependent alteration. With an increasing severity of CHF, the endothelial vascular function deteriorates. In rats with early stages of heart failure, vascular endothelial function was preserved, whereas in more severe stages an impairment was noted (13). Circulating cytokines such as tumor necrosis factor-α (TNF-α) are elevated in severe heart failure. TNF-α impairs the stimulated release of endothelium-derived relaxing factor (EDRF). Plasma levels of TNF in patients with heart failure are correlated with the degree of endothelial dysfunction assessed by infusion of acetylcholine (14). Chronically decreased perfusion of skeletal muscle, increased angiotensin-converting enzyme (ACE) activity in tissues, increased formation of oxygen free radicals, and increased levels of endothelial vasoconstrictive agents are other potential mechanisms involved in the development of endothelial dysfunction in these patients.
In conjunction with endothelial dysfunction, experimental evidence demonstrates a dysfunction of the vascular smooth muscle in heart failure. Zelis et al. (15,16,17), in a series of elegant studies, demonstrated that fluid and sodium retention can impair arteriolar vasodilation in humans. Using a canine model of heart failure produced by rapid ventricular pacing, Zelis et al. (16) also measured the arterial sodium content in the aorta and femoral artery in animals with heart failure and in controls. A significant increase in arterial sodium content was demonstrated in the animals with heart failure. Zelis et al. postulated that arteriolar stiffness as a consequence of increased salt and water content results in an abnormal vasodilation response in heart failure.
During exercise, peripheral vasoconstriction is increased to prevent systemic hypotension in patients with heart failure, given their blunted rise in cardiac output (18). Tissue hypoxia and enhanced sympathetic and angiotensin activation are two proposed mechanisms mediating abnormal peripheral vasoconstriction in CHF (19). Institution of sympathetic and renin-angiotensin blockade does not completely reverse the peripheral derangements seen in CHF, although it modifies it.
Regional specificity of the changes in the peripheral circulation has been demonstrated in patients with CHF. Comparison of peak reactive hyperemia in the forearm and calf musculature of these patients revealed diminished hyperemia only in the leg (20). Correlations with peak VO2 were observed only with peak reactive hyperemia in the calf. Selective deconditioning may be responsible for these regional differences.
Skeletal Muscle Changes in Heart Failure
The metabolic behavior of limb muscles during exercise in patients with heart failure has been examined. Abnormal skeletal muscle metabolism (i.e., reduced oxidative metabolism with earlier shift to glycolytic metabolism) has been demonstrated in patients with heart failure by means of phosphorus 31 magnetic resonance spectroscopy (31P-MRS) (21,22,23,24,25,26,27). These abnormalities appear to be independent of total limb perfusion (22,26,27), histochemical changes (24), muscle mass (25), or severe tissue hypoxia (28).
The kinetics of recovery of skeletal muscle oxygenation are prolonged. At comparable levels of exercise, both VO2 kinetics and muscle oxygenation kinetics were prolonged during recovery in the CHF patients, and the delay worsened with increasing cardiac dysfunction as assessed by peak VO2. A significant correlation between VO2 recovery kinetics and metabolic changes assessed with magnetic resonance spectroscopy has been described (29).
A variety of histochemical changes have been reported in muscle biopsy studies from patients with heart failure. These include fiber atrophy, a decrease in oxidative enzymes, and a shift in fiber composition of the muscle with a significant decrease in fatigue resistant oxidative type I fibers and significant increase in the percentage of glycolytic, fast-twitch, easily fatigable type IIb fibers. Mito-chondrial changes, including a decrease in the volume and surface density of cristae, were described by Drexler et al. (30). The vascularity of skeletal muscles has been assessed by means of capillary-to-fiber ratios, with some investigators reporting a decrease in all measures of capillary density (30,31) and others reporting no change from normal (32).
Additional histochemical evidence of skeletal muscle change is provided by the demonstration of apoptosis in 47% of skeletal muscle biopsies from patients with CHF versus none in healthy volunteers. Peak oxygen consumption was significantly reduced in patients with apoptosis as compared to patients without (12.0 ± 3.7 mL/kg per minute versus 18.2 ± 4.4 mL/kg per minute; p = 0.0005). Apoptosis-positive patients were also found to have a significantly longer history of illness. Intriguingly, a higher level of inducible nitric oxide synthase (iNOS) expression and a lower level of the oncogene bcl-2 expression were also demonstrated in apoptosis-positive biopsies, suggesting possible regulatory mechanisms of apoptosis in the skeletal muscles of these patients. Apoptosis in skeletal
muscles may thus contribute to the reduced exercise capacity in CHF patients (33).
muscles may thus contribute to the reduced exercise capacity in CHF patients (33).
To further clarify the role of skeletal muscle iNOS, as well as reduced phosphocreatine resynthesis in exercise intolerance in CHF, vastus lateralis muscle biopsies and bicycle cardiopulmonary tests were compared between 38 male CHF patients and 8 age-matched controls. The investigators found iNOS expression to be significantly higher in CHF patients. This increased iNOS expression correlated with increased nitric oxide production. Mitochondrial creatine kinase (mi-CK), a key enzyme for transfer of high-energy phosphates, was found to be significantly reduced in CHF patients. Importantly, iNOS expression was negatively correlated with maximal oxygen uptake and mi-CK expression. Taken together, the study findings suggest that iNOS expression may attenuate mitochondrial energy transfer in the skeletal muscle of CHF patients, thereby contributing to the metabolic derangements seen in the myocytes and resulting in the characteristically decreased exercise capacity and increased fatigue in this group of patients (34).
In addition to histochemical changes, patients also exhibit generalized muscle atrophy. Anthropomorphic measurements performed in 62 heart failure patients demonstrated evidence of significant muscle loss in 60% of those patients. Muscle volume assessed using magnetic resonance imaging and muscle mass measurements using DEXA scans revealed reduced muscle volume and mass in patients with heart failure compared to normal subjects (35).
This reduction in muscle mass impacts on peak VO2. Jondeau et al. (36) contrasted peak VO2 during combined upper arm and maximal leg exercise. In normal subjects the addition of more muscle mass via arm exercise did not increase peak exercise performance and VO2 remained unchanged. Thus, cardiac-output response to exercise, rather than amount of exercising muscle, appears to determine peak VO2. However, in patients with severe CHF the addition of upper arm exercise significantly increased peak VO2. The importance of skeletal muscle mass in limiting peak VO2 is suggested by this study.
Intrinsic skeletal muscle changes may represent the primary determinant of exercise performance in a subset of patients with heart failure. Wilson et al. (37) measured exercise hemodynamics and leg blood flow in 34 patients with heart failure and 6 normal subjects. All patients exhibited a reduced exercise capacity, with a peak VO2 of less than 18 mL/kg per minute; however, approximately 25% had normal leg blood flow. Despite normal leg blood flow, lactate release was abnormal in these patients, which implies that their exercise limitation is most probably a consequence of intrinsic skeletal muscle changes rather than a limited cardiac-output response.
In summary, exercise capacity in patients with heart failure is limited not only by changes that affect the ability to increase cardiac output but also by changes in the blood vessels, muscles, and lungs that are a consequence of their disease (Fig. 38-2).
Safety of Exercise Testing
Bed rest was formerly a recommended therapy for heart failure, with physical activity discouraged in this patient pop ulation. Exercise testing was not used in the diagnosis and management of patients with heart failure because of safety concerns. In the Veterans Heart Failure Trial (VHeFT-1) (38), low-level exercise was used to test 607 patients with moderately advanced heart failure (mean peak VO2, 14.5 ± 3.9 mL/kg per minute). This was the first large, multicenter trial to use metabolic stress testing to assess disease severity and response to therapy. Patients underwent two graded, symptom-limited tests of bicycle exercise. The safety of exercise testing in patients with moderate to severe heart failure was demonstrated in that the initial stress test was terminated in only 10 patients (1.6%) for arrhythmias, and in only one patient for hypotension (38). Holter monitoring during and for a period of 4 hours after exercise revealed a 5.7% prevalence of asymptomatic ventricular tachycardia during exercise and a 28.8% prevalence during the rest of the monitoring period. The low incidence of adverse events associated with exercise stress testing in patients with CHF demonstrates that those whose condition is stable can safely exercise in a well-supervised setting.
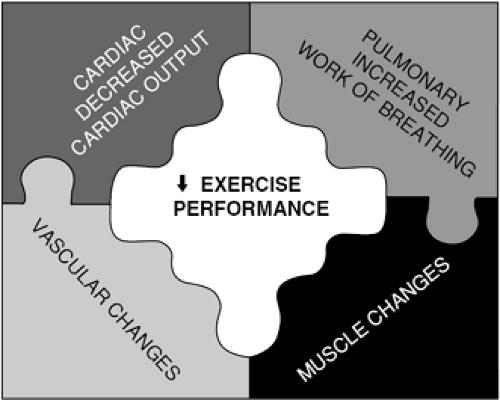 Figure 38-2 Exercise limitations in heart failure arise from a complex interplay of factors, including decreased cardiac output and disease-related changes in the skeletal muscles, peripheral vasculature, and lungs. |
The safety of repeated exercise in CHF was demonstrated in several small training studies of patients with a reduced left ventricular ejection fraction (LVEF). Subsequent exercise testing (39) and exercise training studies in patients with mild to moderate heart failure symptoms also demonstrated no significant ischemic or arrhythmic complications during training.
Prognostic Implications of Exercise Testing
Measurement of exercise peak VO2 has been found to be extremely useful in the risk stratification of patients with heart failure. Indeed, use of peak VO2 in candidate selection for cardiac transplantation has become standard practice. In a prospective study of 114 ambulatory patients with CHF referred to the University of Pennsylvania for cardiac transplantation, a VO2 of less than 14 mL/kg per minute was used as a criterion for acceptance for cardiac transplantation (40). Patients were divided into three groups based
on the results of their cardiopulmonary stress tests. Patients with a peak VO2 below 14 mL/kg per minute were accepted as transplant candidates whereas transplant was deferred for patients with a peak VO2 above 14 mL/kg per minute. A third group included patients with a peak VO2 below 14 mL/kg per minute who had a significant comorbidity that precluded transplant. Age, LVEF, and resting hemodynamic parameters were similar. One-year survival was 94% in patients with a VO2 above 14 mL/kg per minute. Accepted transplant candidates with a VO2 below 14 mL/kg per minute had a 1-year survival of 70%, whereas the patients with a significant comorbidity and reduced VO2 had a 1-year survival of 47% (Fig. 38-3). Patients accepted for transplant had a falsely elevated survival because all transplants were treated as a censored observation. If urgent transplant was counted as death, the 1-year survival fell to 48%. With this approach, we were able to identify the candidates whose transplant could be deferred. We subsequently developed a predictive model to stratify ambulatory transplant candidates according to risk. Peak VO2 was a key parameter in the derivation of the survival score (41). Several recent studies have reaffirmed the prognostic utility of VO2 in the era of beta-blockade and biventricular pacing (42,43).
on the results of their cardiopulmonary stress tests. Patients with a peak VO2 below 14 mL/kg per minute were accepted as transplant candidates whereas transplant was deferred for patients with a peak VO2 above 14 mL/kg per minute. A third group included patients with a peak VO2 below 14 mL/kg per minute who had a significant comorbidity that precluded transplant. Age, LVEF, and resting hemodynamic parameters were similar. One-year survival was 94% in patients with a VO2 above 14 mL/kg per minute. Accepted transplant candidates with a VO2 below 14 mL/kg per minute had a 1-year survival of 70%, whereas the patients with a significant comorbidity and reduced VO2 had a 1-year survival of 47% (Fig. 38-3). Patients accepted for transplant had a falsely elevated survival because all transplants were treated as a censored observation. If urgent transplant was counted as death, the 1-year survival fell to 48%. With this approach, we were able to identify the candidates whose transplant could be deferred. We subsequently developed a predictive model to stratify ambulatory transplant candidates according to risk. Peak VO2 was a key parameter in the derivation of the survival score (41). Several recent studies have reaffirmed the prognostic utility of VO2 in the era of beta-blockade and biventricular pacing (42,43).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


