Fig. 4.1
Group specific prevalence of pulmonary hypertension
Temporal trends in mortality and hospitalization where PH was listed as any contributing cause of death or any-listed hospital diagnosis in the National Vital Statistic System and hospital discharge data from the National Hospital Discharge Survey for 1980–2002 show stability in PH-related mortality (per 100,000: 5.2 in 1980 to 5.4 in 2002), but an increase in hospitalizations (per 100,000: 40.8 in 1980 to 90.1 in 2002) over the same two decade time period [13]. However, the mortality rates increased among women but decreased among men during this period. In Medicare enrollees aged >65 years, the annual number of hospitalizations for pulmonary hypertension as any-listed diagnosis tripled from 1990 to 2002. The age-standardized hospitalization rate per 100,000 in this group was 197.8 in 1990 and 649.7 in 2002 [13]. Since these data are dependent on appropriate ICD coding, it is unclear if the increase is due to an increased awareness of the diagnosis versus an actual increase in the number of incident PH cases. However, ageing and an attendant increase in the burden of heart failure in the general population suggests that PH might continue to be a more frequent diagnosis associated with hospitalizations and mortality.
Pulmonary Hypertension: Demographics and Comorbidities
Age
The PASP and prevalence of PH increases with age [2, 3, 14]. This relationship is independent of other comorbidities and cardiopulmonary function [3]. The prevalence ratio of having PH in subjects >65 years of age is tenfold higher compared to those younger than 45 years of age. The higher prevalence of PH in older people may be related to decreasing vascular compliance of the pulmonary arteries with age, which also occurs in the systemic arteries [2, 3].
Sex
Studies reporting on the relationship between PASP and PH and sex in the general population have yielded conflicting results. In the AA population, women have an increased age-adjusted prevalence of PH compared to men that is independent of other comorbidities. Fifteen percent of AA women over 65 years of age have PH compared to 9 % of AA men [3] (Fig. 4.2). In contrast, studies performed in a general population of young adults [15], and in normal echocardiograms performed over a decade at Massachusetts General Hospital [14] showed higher PASP in males, while in the Olmstead County population cohort [2] no relationship between sex and PA pressure was observed. Determining whether the relationship between sex and PH is race-specific and relates to specific biologic factors such as estrogen and estrogen metabolites needs further validation in additional population-based cohorts [16–20].
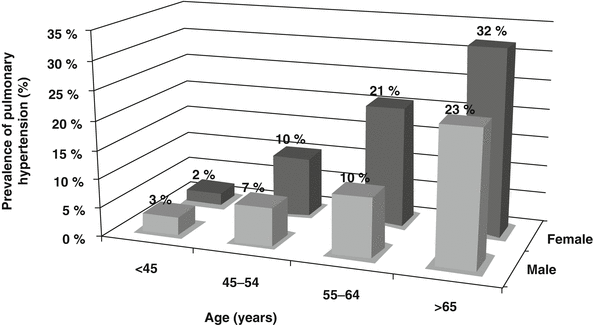
Fig. 4.2
Prevalence of pulmonary hypertension in African-American men and women across age groups [3]
Race/Ethnicity
Little data is available comparing PASP or PH prevalence according to race. In the CARDIA (Coronary Artery Risk Development in Young Adults) study, AAs were found to have higher mPAP (assessed by pulmonary artery acceleration time on echocardiography) independent of age, sex, body mass index (BMI), smoking status, and presence of diastolic dysfunction [15]. Similarly, in one community-based cohort, PH prevalence was 6.8 % in AA compared to an estimated prevalence of ~2–3 % in whites based on the cumulative frequency of PASP [2, 3]. However, these cohorts had differences in cardiopulmonary co-morbidities, and it remains to be determined if race is an independent risk factor for developing PH after adjusting for age and other chronic conditions.
Obesity
Obesity or higher BMI is positively associated with PH prevalence and PASP [2, 3, 14, 15]. For example, in AA, the presence of obesity increases the adjusted prevalence of PH by 66 % compared to individuals with normal BMI [3]. A similar relationship between obesity and PASP > 30 mmHg (odds ratio of 1.67) was observed by McQuillan and colleagues [14]. The pathophysiological mechanisms are not delineated clearly, but may be related to the presence of left ventricular diastolic dysfunction, sleep disordered breathing, obesity-hypoventilation syndrome or insulin resistance/adipose leading to vascular inflammation.
Cardiovascular Comorbidities
Considering that the most common cause of PH is related to left ventricular (LV) dysfunction and left atrial (LA) hypertension, it is not surprising that the presence of PH is related to changes in LV systolic and diastolic function and left sided valvular diseases. Hypertension, coronary artery disease, diabetes, congestive heart failure and severe mitral and aortic valvular disease are more common in subjects with PH [2, 3]. The underlying mechanism(s) to account for these relationships are likely related to presence of diastolic dysfunction and elevated LA pressures. Indeed, echocardiographic markers of systolic dysfunction and LV diastolic function (LV hypertrophy, LA size and Doppler indices of diastolic function) are correlated with PASP and PH [2, 3, 14, 15].
Pulmonary Comorbidities
COPD is associated with PH [21] and the underlying mechanism(s) are multifactorial. Hypoxia, vascular remodeling, destruction of capillaries in emphysema, hyperinflation, and LV diastolic dysfunction constitute likely mechanisms responsible for increased PASP in the settings of parenchymal lung diseases. The prevalence of chronic lung diseases increases with increasing PASP in the general population [2, 3]. Also, airway obstruction assessed by spirometry is associated with a twofold increase in the adjusted risk for PH independent of other co-morbidities [3]. A significant association between a restrictive spirometry pattern [22–26] and the presence of PH was also observed in the Jackson Heart Study Cohort [3]. A restrictive spirometry pattern can be caused by fibrotic lung disease, obesity, diabetes, heart disease, and hypertension [22, 25]. However, the relationship between restriction and PH was independent of these co-morbidities suggesting other as yet unrecognized factors contribute to these associations.
Pulmonary Hypertension: Prognosis
Elevated PASP has been shown to result in higher mortality in the general population. This was demonstrated in the both Caucasian (Adjusted HR: 1.46 per 10 mmHg) [2] and AA cohorts (Adjusted HR: 1.38 per 10 mmHg) [27]. Also, elevated PASP is an independent predictor of admissions for congestive heart failure in the general population (Adjusted HR: 2.03 per 10 mmHg). Higher mortality has been noted in patient cohorts with multiple cardiopulmonary morbidities such as the U.S. military Veteran population in which 44.1 % of patients with echocardiographically-determined PASP > 60 mmHg were deceased at a median follow up of 832 days [9].
World Health Organization Group 1 Pulmonary Arterial Hypertension
The epidemiology of WHO Group 1 PAH is the most well defined among the five WHO groups. The earliest prospective multicenter registry from the National Institutes of Health, which included patients with idiopathic PAH (IPAH), heritable PAH (HPAH), and PAH associated with anorexigen use, reported a mean age of 36 ± 15 years and a female:male ratio of 1.7:1 overall (Table 4.1). Before the advent of targeted PAH therapy, 1-, 3-, and 5-year survival for this cohort was 68 %, 48 %, and 34 %, respectively, with an estimated median survival of 2.8 years (95 % confidence interval 1.9–3.7 years) [28] (Table 4.2). This early snapshot of what was then known as “primary pulmonary hypertension”, a rare disease affecting young females of child-bearing age, has evolved in recent years as we have come to understand more about WHO Group 1 disease including PAH associated with conditions such as connective tissue disease (CTD).
Table 4.1
Major PAH registries and characteristics of patients enrolled
Registry | Years | Type | N | Age, yrs | Female sex, % | FC III/IV, % | 6MWD, m | RAP, mmHg | mPAP, mmHg | CI, L/min/m2 | PVRI (mmHg/L/min∙m2) or PVR (Wood units) |
|---|---|---|---|---|---|---|---|---|---|---|---|
1981–1985 | IPAH, HPAH anorexigen | 187 | 36 ± 15 | 1:7:1 | – | – | 10 ± 6 | 60 ± 18 | 2.3 ± 0.9 | 26 ± 14 | |
PHC [30] | 1982–2007 | Mixed, 42 % IPAH | 576 | 48 ± 14 | 77 | 80 | – | 11 ± 6 | 52 ± 14 | 2.2 ± 0.9 | 12 ± 7 |
SMR [31] | 1986–2001 | Mixed, 47 % IPAH | 374 | – | 70 | – | – | – | – | – | – |
Mayo [32] | 1995–2004 | Mixed, 56 % IPAH | 484 | 52 ± 15 | 75 | 71 | 329 ± 125 | 13 ± 6 | 53 ± 13 | 2.5 ± 0.8 | – |
Chinese [33] | 1999–2004 | IPAH, HPAH | 72 | 36 ± 12 | 71 | 61 | – | 8 ± 3 | 59 ± 15 | – | 20 ± 9 |
UK/Ireland [34] | 2001–2009 | Mixed, 93 % IPAH | 482 | 50 ± 17 | 70 | 84 | 292 ± 123 | 10 ± 6 | 54 ± 14 | 2.1 ± 0.7 | 13 ± 6 |
French [35] | 2002–2003 | Mixed, 39 % IPAH | 674 | 50 ± 15 | 65 | 75 | 329 ± 109 | 8 ± 5 | 55 ± 15 | 2.5 ± 0.8 | 21 ± 10 |
REVEAL [36] | 2006–2007 | Mixed, 46 % IPAH | 2525 | 53 ± 14 | 80 | 51 | 366 ± 126 | 9 ± 6 | 51 ± 14 | 2.4 ± 0.8 | 21 ± 13 |
COMPERA [37] | 2007– | Mixed, 62 % IPAH | 1283 | 68 (55–75) | 64 | 87 | 303 ± 132 | 8 ± 5 | 44 ± 12 | 2.3 ± 0.8 | 10 ± 6 |
Spain [38] | 2007–2008 | Mixed, 36 % IPAH | 866 | 45 ± 17 | 71 | 69 | 363 ± 120 | 9 ± 5 | 54 ± 16 | 2.6 ± 0.9 | 12 ± 6 |
Chinese [39] | 2007–2009 | 63 % IPAH, 37 % CTD | 276 | 33 ± 15a | 70a | 52a | 394 ± 114a | 12 ± 6a | 63 ± 18a | 2.5 ± 0.9a | 17 ± 10a |
Table 4.2
Survival rates at 1, 3, and 5 years among patients enrolled in registries
Registry | Years | 1 year, % | 3 year, % | 5 year, % |
|---|---|---|---|---|
NIH [28] | 1981–1985 | 68 | 48 | 34 |
PHC [40] | 1982–2007 | 86 | 69 | 61 |
Mayo [32] | 1995–2004 | 81 | 61 | 48 |
Chinese [33] | 1999–2004 | 68 | 39 | 21 |
UK/Ireland [34] | 2001–2009 | 93 | 73 | 61 |
French [41] | 2002–2003 | 83 | 67 | 58 |
REVEAL [42] | 2006–2007 | 91 | 74 | 65 |
COMPERA [43] | 2007 | 93 | 84b | – |
Spain [38] | 2007–2008 | 89 | 77 | 66a |
Chinese [39] | 2007–2009 | 92 | 80 | 75a |
Prevalence and Incidence
A number of population-based registries have offered insight into PAH prevalence and incidence. In several countries in Europe, the centralization of PAH care has allowed for wide-scale estimates of pulmonary vascular disease burden. The network of French centers (n = 17) described a prevalence of 15 cases/million for WHO Group 1 PAH (5.9 cases/million for IPAH), although significant regional variation was noted (5–25 cases/million French adults), and an incidence of 2.4 cases/million/year in 2002–2003 [35]. The prevalence of IPAH, HPAH, and anorexigen-associated PAH was 6.6 cases/million in 2009, with an estimated incidence of 1.1 cases/million/year in the United Kingdom (UK) and Ireland [34]. Across Spain, the prevalence of PAH has been reported to be 16 cases per million and the incidence 3.7 cases/million/year [38]. The overall population prevalence of PAH in Scotland was 52 cases per million population; prevalence estimates were higher when compared to an expert referral center (the Scottish Pulmonary Vascular Unit), raising the question of whether registries from expert centers adequately capture the true population impact of pulmonary vascular disease. The overall annual incidence of PAH over a 16 year study period as estimated from the Scottish Morbidity Record was 7.1 cases per million population; the incidence of IPAH, CTD-associated PAH and congenital heart disease (CHD) associated PAH respectively was 3.3, 2.1 and 1.7 cases per million population [31].
Survival
Multiple registries have captured survival in both the pre- and post-PAH treatment eras (Table 4.2) [28, 32–34, 38–43]. Comparing these estimates, it appears that short-term survival has improved over time and is approximately 90 % at 1 year and 75 % at 3 years. Longer-term survival remains poor, however, with registries reporting survival rates between 21 and 75 % at 5 years. Of course, registries are susceptible to survivor bias and outcome estimates may be heavily influenced by mixing of prevalent versus incident cases.
Demographics: Age, Sex, and Race
Single center US-based registries including various WHO Group 1 etiologies have reported slightly older average age (4th–5th decade of life) than that reported in the NIH registry, but confirmed 70 % female predominance, as have other national reports [30–32, 35, 40]. In 2006, the Registry to Evaluate Early And Long-term pulmonary arterial hypertension disease management (REVEAL) began enrolling patients with WHO Group 1 PAH as traditionally defined as well as patients with PH and elevated left heart filling pressures (pulmonary capillary wedge pressure 16–18 mmHg) from 54 US-based community and academic sites [36, 44]. In patients with IPAH, the average age at enrollment was 53 ± 15 years of age and 80 % were female, signaling a possible shift in demographics as compared to earlier PAH registries.
Whether the observations from REVEAL signal a true change in disease biology (e.g., via epigenetic influences), greater disease awareness in an aging population, and/or that the “pure” idiopathic PAH pathophenotype has been diluted to include overlapping forms of PH in modern clinical practice is not known. Similar trends were observed in the UK and Ireland registry, where patients enrolled later (2007–2009) tended to be older, more obese, and have more comorbid diabetes and ischemic heart disease as compared to PAH patients enrolled earlier (2001–2003) [34]. The Comparative Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) database, which commenced in 2007 and included patients from multinational European centers, reported an average age at diagnosis of 71 ± 16 years among incident IPAH cases [37]. Patients diagnosed at a younger age appear to have worse hemodynamic burden, yet better functional tolerance and survival as compared to older aged patients, who have more comorbidities and are more commonly obese [34, 37]. In addition, survival with modern PAH treatment had improved. This has been shown in a retrospective evaluation of cohort of patients from 2007 to 2009 in China, where targeted PAH therapy was introduced in 2006 [39]. While baseline characteristics for incident IPAH patients remained similar to the NIH registry, survival with modern PAH treatment had improved, as has been observed in other cohorts [45].
Female sex has long been established as the major clinical risk factor for PAH, but recent epidemiologic data has suggested that males have poorer survival than females and that age may be an important modifier of the relationship between sex and outcomes [13, 38, 41–43, 46]. Lifetime hormonal fluctuations (e.g., menarche, menopause, androgen fluctuations at puberty and in older age) that differentially regulate cardiopulmonary function as well as sex-specific responses to therapy have been proposed as possible explanations for these observations [20, 47–49]. In fact, among older patients, sex-based differences in PAH prevalence appear to be diminished [34, 37, 46]. Males have greater hemodynamic burden at diagnosis as compared to females, but these differences are attenuated after age 45 in a cohort of clinical trial participants [46]. Similarly, in the REVEAL registry, men had higher RAP and mPAP at diagnosis (as well as worse survival especially in those older than 60 years of age) [42, 50]. These observations have not been consistent across all registries, however, and further work is needed to refine the sex-age interaction in pulmonary vascular disease [34, 39].
Less is known about the role of race/ethnicity in disease prevalence and outcomes. In the NIH registry, the distribution of race/ethnicity observed in patients was similar to that of the general US population, although there was a greater female:male predominance among blacks (4.3:1) [28]. This observation was also made in the UK and Ireland, where 12.3 % of 424 patients were nonwhite, 85 % of whom were female (as compared to 70 % female in white patients) and in the US, where a 5.4:1 female to male ratio was observed in AA from the REVEAL registry [34, 36]. AA have higher rates of hospitalization and death as compared to whites, and appear to have a less robust response to treatment with endothelin receptor antagonists in PAH clinical trials [13, 47, 51, 52]. Han Chinese patients appear to have similar characteristics to white patients at baseline; in a small cohort from a single US center, Asian descent was an independent predictor of death [33, 52]. While neither sex nor race/ethnicity were associated with delay in diagnosis (>2 years between symptom onset and evaluation for PAH) in the REVEAL registry, it is unknown whether these possible differences in outcomes are due to true differences in disease biology, variable therapeutic responses, and/or confounding by socioeconomic status such as limited access to care as has been noted in other cardiopulmonary diseases [53]. Irrespective of the precise reason, it is important to note that lower socioeconomic status (assessed as a composite of level of education, medical reimbursement, employment status, and household income) was strongly associated with mortality after adjustment for age, sex, disease factors, and PAH treatments in the Chinese cohort [54].
Group 1 Pulmonary Arterial Hypertension Subtypes and Associated Conditions
Heritable Pulmonary Hypertension
Mutations in the bone morphogenic protein receptor type 2 (BMPR2), a member of the transforming growth factor [TGF]-β family, are present in 70–80 % of families with PAH and roughly 25 % of patients with IPAH [55, 56]. These mutations are transmitted in an autosomal dominant fashion with incomplete penetrance. TGF-β signaling including the BMPR2 pathway is felt to play a major role in disease pathogenesis. Further evidence is provided by rarer mutations in TGF-β receptor components activin-like receptor kinase-1 (ALK-1) and endoglin (ENG), both of which have been implicated in hereditary hemorrhagic telangiectasia and PAH [57, 58]. Variants in other genes, including mothers against decapentaplegic homologue (SMADs), proteins in the TGF-β signaling pathway, and more recently caviolin 1 (CAV1) and the potassium channel subfamily K member 3 (KCNK3) (neither of which directly relate to TGF-β) have also been implicated in heritable disease although are less common than BMPR2 mutations [59–62]. BMPR2 mutation carriers tend to present at a younger age and with more severe hemodynamic compromise and ALK-1 carriers may have a worse overall prognosis as compared to other idiopathic and heritable patients [63, 64]. Patients with HPAH otherwise have similar features to those with IPAH, and as such many registries and clinical trials have approached these two entities as one sub-group.
Drug- and Toxin-Induced Pulmonary Hypertension
A number of drugs and toxins have been implicated in the development of pulmonary vascular disease, some with more definitive epidemiologic links than others [65]. A comprehensive list and ranking of definite, possible, likely, and unlikely agents was updated during the 5th World Symposium in Nice, France in 2013 [65]. The most classic example of drug-induced PAH are the anorexigens such as fenfluramine, in which risk appears to be exposure duration-dependent (particularly beyond several months), although fatal cases have been reported from just short-term use [66–72]. Fenfluramine products were withdrawn from the market worldwide in the 1990s, but a related available compound, benfluorex, has been linked to PH as well as to valvular disease [71]. Although tyrosine kinase inhibitors have been studied to treat PAH, the use of dasatinib for chronic myelogenous leukemia has been associated with the development of PAH [73–75]. Treatment with interferon has also been identified as a possible risk factor after case reports emerged of patients treated with interferon for hepatitis C, multiple sclerosis, and oncologic conditions developed PAH [65, 76–81]. Drug- and toxin-associated PAH accounted for 2 % of PAH patients in the UK and Ireland, 7 % of incident and 13 % of prevalent cases in France, and 11 % of patients in REVEAL, with similar baseline characteristics to those patients with IPAH [36, 41, 82].
Connective Tissue Disease
A number of systemic diseases are associated with the development of pulmonary vasculopathy, although the mechanisms by which PAH develops in these varied conditions are generally poorly understood. In patients with CTD, those with systemic sclerosis are at greatest risk. Roughly 12 % of CTD patients develop PAH and it is a major cause of death in systemic sclerosis [83–85]. To that end, current recommendations are to screen patients with systemic sclerosis or scleroderma spectrum disorders for PAH using noninvasive testing (echocardiogram, diffusion capacity of the lungs for carbon monoxide) and biomarkers (N-terminal pro-brain natriuretic peptide) annually [86, 87] (Table 4.3). Additional CTDs such as systemic lupus erythematosus, mixed connective tissue disease, and rheumatoid arthritis have been linked to pulmonary vascular disease, and while the true prevalence of PAH in these conditions is unknown PAH appears to occur less commonly and is associated with better outcomes than when associated with systemic sclerosis [101, 102].
Table 4.3
Conditions associated with pulmonary hypertension and considerations for screening
WHO group | Associated condition | Estimated prevalence, % | Screening |
|---|---|---|---|
I. | Systemic sclerosis and scleroderma spectrum disorders | ||
Human immunodeficiency virus infection | None recommended | ||
Portopulmonary hypertension | Pre-transplant evaluation | ||
Congenital heart disease | At diagnosis | ||
V. | Sickle cell disease | 10 % PAH, higher using tricuspid jet velocity [99] | Adults > 18 years of age, echocardiography every 1–3 years [100] |
After IPAH, CTD-PAH patients tend to be the second most represented subgroup in registries, allowing for comparisons between these groups [31, 35, 36, 38]. CTD-PAH patients tend to be older, have less hemodynamic impairment, and may have left-sided abnormalities such as left atrial enlargement and increased left-ventricular end-diastolic diameter, signaling the potential role of pulmonary venous hypertension in some patients [36, 39]. Survival is poorer in CTD-PAH as compared to idiopathic disease and, among CTD-PAH, lowest in patients with systemic sclerosis [29, 39, 102, 103].
Human Immunodeficiency Virus
Non-rheumatologic systemic diseases that are associated with the development of PAH include human immunodeficiency virus (HIV) infection [65, 104]. Over the past several decades, multiple studies have estimated the prevalence of pulmonary vascular disease in HIV infected patients to be approximately 0.5 % [88–90]. While rare, PAH complicating HIV infection has not decreased appreciably in frequency despite the advent of antiretroviral therapy, and the mechanistic link between PAH and HIV has not been clearly established [89]. HIV ribonucleic acid has not been isolated in the pulmonary vasculature, but HIV proteins such as Tat and Nef cause endothelial dysfunction [105–111]. Disease characteristics are similar to IPAH patients, although female sex does not appear to be a risk factor for the development of PAH in HIV [35, 112]. While HIV-associated PAH has historically had the least favorable prognosis among PAH subtypes, patients may respond well to targeted PAH therapy and contemporary survival rates compared to those with IPAH (88 % at 1 year and 72 % at 3 years) [38, 113, 114].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


