Definition
Hemodynamic characteristics
Pulmonary hypertension
mPAP ≥ 25 mmHg
Pre-capillary PH
mPAP ≥ 25 mmHg
PAOP ≤ 15 mmHg
CO normal or reduced
Post-capillary PH
mPAP ≥ 25 mmHg
PAOP > 15 mmHg
Combined post-capillary and pre-capillary PH
mPAP ≥ 25 mmHg
PAOP > 15 mmHg
Diastolic PAP—PAOP > 7 mmHg
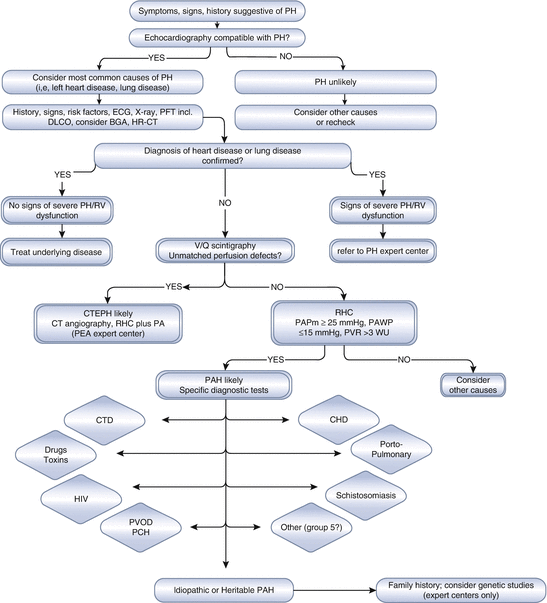
Classification
The first classification of PH was described in 1973 and categorized patients as having “primary” or “secondary” pulmonary hypertension according to the presence or absence of an identifiable cause for the disease. During the second world symposium on PH, in 1998, the basis for the classification system currently used was proposed. This system groups patients with similar pathological findings, similar hemodynamic profiles, as well as similar management strategies into a single category. Five different categories were proposed: (1) pulmonary arterial hypertension; (2) pulmonary hypertension due to left heart disease; (3) pulmonary hypertension due to chronic lung disease and/or hypoxia; (4) chronic thromboembolic pulmonary hypertension; and (5) pulmonary hypertension due to unclear multifactorial mechanisms (previously called “miscellanea”) [8]. Although small modifications have been made during the last decade, the concept of the current classification remains the same. The updated classification for pulmonary hypertension, derived from the last world symposium on PH, is presented in Table 2.2 [9].
Table 2.2
Clinical classification of pulmonary hypertension 9
1. Pulmonary arterial hypertension (PAH) |
1.1. Idiopathic PAH |
1.2. Heritable |
• 1.2.1. BMPR2 |
• 1.2.2. ALK1, ENG, SMAD9, CAV1, KCNK3 |
• 1.2.3. Unknown |
1.3. Drug- and toxin-induced |
1.4. Associated with |
• 1.4.1. Connective tissue diseases |
• 1.4.2. HIV infection |
• 1.4.3. Portal hypertension |
• 1.4.4. Congenital heart diseases |
• 1.4.5. Schistosomiasis |
1′ Pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis |
1″ Persistent pulmonary hypertension of the newborn |
2. Pulmonary hypertension owing to left heart disease |
• 2.1. Left ventricular systolic dysfunction |
• 2.2. Left ventricular diastolic dysfunction |
• 2.3. Valvular disease |
• 2.4. Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies |
3. Pulmonary hypertension owing to lung diseases and/or hypoxia |
• 3.1. Chronic obstructive pulmonary disease |
• 3.2. Interstitial lung disease |
• 3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern |
• 3.4. Sleep-disordered breathing |
• 3.5. Alveolar hypoventilation disorders |
• 3.6. Chronic exposure to high altitude |
• 3.7. Developmental lung diseases |
4. Chronic thromboembolic pulmonary hypertension (CTEPH) |
5. Pulmonary hypertension with unclear multifactorial mechanisms |
• 5.1. Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy |
• 5.2. Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis: lymphangioleiomyomatosis, neurofibromatosis, vasculitis |
• 5.3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders |
• 5.4. Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental PH |
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) encompasses a group of clinical conditions that result in precapillary PH and share similar pathological and/or clinical findings. Idiopathic PAH corresponds to sporadic disease in which no family history of PAH or an identified risk factor is present [8]. IPAH is only diagnosed after alternative diagnoses are ruled out according to current guidelines [1]. Up to 70 % of familial cases of PAH have been linked to germline mutations in the gene coding for the bone morphogenetic protein receptor 2 (BMPR2), a member of the transforming growth factor beta (TGF-β) signaling family [10]. BMPR2 mutations have also been detected in a significant proportion of apparently idiopathic cases without familial history [11]. Indeed, the distinction between idiopathic and familial PAH with BMPR2 mutations is artificial, as all patients with a BMPR2 mutation have heritable disease. Thus, it was decided to abandon the term “familial PAH” in favor of the term “heritable PAH” [8]. Heritable forms of PAH include those with identified mutations (mainly BMPR2 but also ACVRL1 or endoglin and more recently SMAD9, KCNK3, and CAV1) and familial cases without identified mutations. Idiopathic and heritable PAH are more common in women than in men with a gender ratio around 2:1 [12] and as high as 3.2 in the REVEAL registry [13].
PAH patients carrying a BMPR2 mutation are prone to be younger at diagnosis and present with more severe disease than IPAH patients without a BMPR2 mutation [14]. As a result, genetic testing and counseling have played an increasingly important role in the comprehensive assessment of patients with newly diagnosed PAH [15–17].
Besides genetic predisposition, there are a number of risk factors that are associated with the development of PAH. The use of aminorexfumarate, a potent appetite suppressant, in the 1960s led to an outbreak of rapidly progressive PAH in Switzerland, Austria, and Germany. The incidence of PAH in patients who had used aminorex was shown to be about 0.2 % with a median exposure-to-onset time of 8 months, as first described in a Swiss medical clinic. The incidence was proportional to the amount of drug taken and if discontinued early enough, a regression of PAH could be seen. Subsequently, aminorex was withdrawn from the market in 1968 [18]. More than 20 years later, fenfluramine and dexfenfluramine were marketed as appetite suppressants with another outbreak of drug-induced PAH associated with these two agents in the 1980s–1990s. PAH cases in patients exposed to fenfluramine derivatives share clinical, functional, hemodynamic, and genetic features with IPAH. This suggests that fenfluramine exposure represents a potential trigger for PAH without influencing its clinical course [19]. The association of fenfluramine and dexfenfluramine with the development of PAH was confirmed by a registry that enrolled 1,335 subjects at tertiary PH centers in the United States between 1998 and 2001. Of note, these agents have also been associated with an increased risk of valvular heart diseases, presumably because of their serotoninergic properties. As a result, fenfluramine and dexfenfluramine were withdrawn from the market in the late 1990s [20].
More recently, Benfluorex, a benzoate ester that shares structural and pharmacologic characteristics with dexfenfluramine and fenfluramine, has also been associated with the development of PAH. The active and common metabolite of each of these molecules is norfenfluramine, which itself has a chemical structure similar to that of the amphetamines. Given its pharmacological properties, benfluorex would be expected to have similar toxic effects to the fenfluramine derivatives [21, 22]. An outbreak of valvular heart disease and/or PAH induced by benfluorex use was identified by the French PH network from June 1999 to March 2011 and included 85 cases of PAH. The analysis of these cases led to the withdrawal of benfluorex from the French market in 2009 [23].
Other classes of drugs have also been linked to the development of PAH. Cases of precapillary PH fulfilling the criteria of drug-induced PAH have been reported in chronic myelogenous leukemia patients treated with the tyrosine kinase inhibitor dasatinib. At diagnosis, patients had moderate to severe precapillary PH with functional and hemodynamic impairment. Clinical, functional, and hemodynamic improvements were observed within a few months of dasatinib discontinuation in most patients. However, after a median follow-up of 9 months (range 3–36 months), the majority of patients failed to demonstrate complete clinical and hemodynamic recovery and no patients reached a normal value of mPAH (≤20 mmHg). The lowest estimate of incident PH occurring in patients exposed to dasatinib in France was 0.45 %. Thus, dasatinib may induce severe PAH, suggesting a direct and specific effect of dasatinib on pulmonary vessels [24].
The potential association of PAH and the use of interferon alfa or beta has also been reported. Fifty-three PAH patients in the French registry had a history of interferon use, raising the question about whether or not a causal relation could be present.
The presence of genetic abnormalities and risk factors (such as specific drug exposures) reinforces the “multiple hit” concept for the development of pulmonary hypertension [25]. The list of the recognized risk factors potentially associated with the development of pulmonary hypertension is presented in Table 2.3.
Table 2.3
Updated classification for drug- and toxin-induced PAHa
Definite | Possible |
|---|---|
Aminorex | Cocaine |
Fenfluramine | Phenylpropanolamine |
Dexfluramine | St. John’s wort |
Toxic rapeseed oil | Chemotherapeutic agents |
Benfluorex | Interferon α and β |
SSRIs b | Amphetamine-like drugs |
Likely | Unlikely |
Amphetamines | Oral contraceptives |
Tryptophan | Estrogen |
Methamphetamines | Cigarette smoking |
Dasatinib |
One of the most important forms of PAH is the one associated with connective tissue diseases, not only due to its clinical course but also because of its prevalence. Connective tissue diseases (CTD) represented about 15 % of all PAH cases in the French registry, with systemic sclerosis and systemic lupus erythematosus as the leading causes (76 and 15 % of all CTD-PAH, respectively) [26]. CTD represented about 25 % of cases in the REVEAL registry from the United States [46]. The prognosis of these patients is worse than other forms of PAH, reaching a 1-year mortality of about 30 % as compared to about 15 % in IPAH [27, 28]. Recently, it has been suggested that implementation of a systematic screening program that allows the use of specific therapies in a less symptomatic phase of the disease might result in better long-term outcome for this subgroup of PAH patients [29].
Patients with HIV infection also have a greater risk of developing PAH. The prevalence of PAH in this group is estimated to be 0.5 %, with clinical and hemodynamic presentation very similar to IPAH [30, 31]. Prognosis of this particular subgroup of PAH has improved in recent years. In the REVEAL registry, the mortality of HIV-PAH patients was 7 and 25 % at 1 and 3 years, respectively [32].
Another group at risk for PAH is patients with portal hypertension. About 6 % of these patients develop PAH [33] independent of the severity of their liver disease, although the long-term prognosis of these patients is related to the severity of both liver and pulmonary vascular diseases [34]. Portopulmonary hypertension represents an important problem for liver transplantation programs since its presence is related to increased mortality during the procedure [35]. The prognosis in POPH is worse than in IPAH. Recent data suggest a 3-year survival of 40 % [36].
About 10 % of children with congenital heart disease (CHD) develop PAH. Since more of these children now survive to adulthood, PAH associated with CHD is a significant subgroup of PH seen in many referral centers [37]. According to the last world symposium, patients with CHD-PAH (except those with more complex congenital heart defects) should be subclassified into four different subgroups (Table 2.4). The concept of this subclassification is to provide guidance on the management of the disease. For instance, patients with small cardiac defects and PAH are considered as having IPAH and coincidental congenital heart disease and should be managed as any other patient with idiopathic disease. On the other hand, patients with CHD and persistent left-to-right shunts should be considered for possible correction of the defect causing the shunt, although criteria for the definition of operability is still a matter of debate [9].
Table 2.4
Updated clinical classification of pulmonary arterial hypertension associated with congenital heart diseasea
1. Eisenmenger syndrome |
Includes all large intra- and extracardiac defects which begin as systemic-to-pulmonary shunts and progress with time to severe elevation of pulmonary vascular resistance (PVR) and to reversal (pulmonary-to-systemic) or bidirectional shunting; cyanosis, secondary erythrocytosis, and multiple organ involvement are usually present. |
2. Left-to-right shunts |
• Correctableb |
• Noncorrectable |
Include moderate to large defects; PVR is mildly to moderately increased, systemic-to-pulmonary shunting is still prevalent, whereas cyanosis is not a feature. |
3. Pulmonary arterial hypertension (PAH) with coincidental congenital heart disease. Marked elevation in PVR in the presence of small cardiac defects, which themselves do not account for the development of elevated PVR; the clinical picture is very similar to idiopathic PAH. To close the defects is contraindicated. |
4. Postoperative PAH |
Congenital heart disease is repaired but PAH either persists immediately after surgery or recurs/develops months or years after surgery in the absence of significant postoperative hemodynamic lesions. The clinical phenotype is often aggressive. |
Schistosomiasis is an infectious disease caused by parasitic trematode worms that is strongly linked to poor sanitary conditions and poverty. Nevertheless, due to migratory practices, the prevalence of schistosomiasis is increasing in nonendemic regions. Pulmonary hypertension represents one of the most severe complications of chronic schistosomiasis [38]. A screening program for pulmonary hypertension in a tertiary center in Brazil identified a 4.6 % prevalence of PAH among patients diagnosed with hepatosplenic Schistosomiasis mansoni [39]. When this prevalence rate is considered in the context of the high prevalence of schistosomal infection globally, schistosomiasis associated PAH (Sch-PAH) is one of the leading causes of PH in the world. Indeed, it is estimated that up to 30 % of all pulmonary hypertension patients followed at referral centers in Brazil have Sch-PAH [40]. A subsequent study showed that although Sch-PAH has a clinical profile similar to IPAH at the time of diagnosis, the clinical course appears to be more benign with a 3-year mortality of about 15 % [41].
Epidemiology
Our knowledge of the epidemiology of PAH has changed dramatically over the last 30 years. The earlier clinical picture of IPAH was mainly derived from the landmark study conducted by the National Institutes of Health (NIH) in the 1980s [42, 43]. By that time, IPAH was described as a disease affecting young patients (mean age of 36 yo), with a female to male ratio of 1.7:1. Time from the first symptom to the appropriate diagnosis exceeded 2 years and the mortality rate at 1 year was about 32 %. Since then, the development of target therapies has not only led to improved survival but has also increased disease awareness. Several multicenter registries have been published in recent years, evidencing a changing epidemiology of PAH (Tables 2.5 and 2.6). The first of these multicenter registries was the French national registry. A total of 674 patients were enrolled by 17 different referral centers [26]. The French Registry confirmed the female predominance, but described a mean age at diagnosis of 50 years, with older patients presenting worse prognosis [27, 45]. Concomitantly, data from the multicenter US registry REVEAL, that included more than 2,000 patients from 54 reference centers, also demonstrated this older age at diagnosis (53 yo) as well as a worse prognosis in male patients presenting at older age [46, 47]. Both registries also demonstrated disturbing data concerning PAH diagnosis. Although an increase in disease awareness is believed to have occurred during the last decade, the appropriate diagnosis still takes about 2 years with most of the patients being diagnosed in functional class III and IV. This is particularly alarming if one takes into consideration that the worse the functional class at the diagnosis, the worse the survival despite all available medical treatments. Therefore, late diagnosis is a true problem, more specifically for IPAH, and might be one of the scenarios to be changed in the near future, in order to better improve survival.
General information of PAH registries from different countries and time periods | ||||||
|---|---|---|---|---|---|---|
Registry (ref. #) | Study cohort | Study design and time period | No. of centers | No. of patients | Incidence/prevalence | Predominant etiologies of PAH |
U.S. NIH | IPAH | Prospective, 1981–1985 | 32 | 187 | NA | NA |
U.S. PHC | Group 1 PH, age >18 years | Retrospective, 1982–2004; prospective, 2004–2006 | 3 | 578 | NA | IPAH, 48 %; CTD-PAH, 30 %; CHD-PAH, 11 % |
Scottish-SMR
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||||