Fig. 12.1
The endothelium is responsible for a number of physiological functions, including (1) regulation of vascular tone, (2) control of blood fluidity and coagulation, and (3) regulation of inflammatory processes. cAMP cyclic adenosine monophosphate, cGMP cyclic guanosine monophosphate, COX cyclooxygenase, BH4 tetrahydrobiopterin, IL interleukin, TNF tumour necrosis factor, l -arg l-arginine, l -cit l-citrulline, NO nitric oxide, NOS nitric oxide synthase, O 2 −superoxide (With permission from Marti et al. [9])
12.2.1 Regulation of Vascular Tone
12.2.1.1 Nitric Oxide
The endothelium regulates vascular tone through the rapid synthesis of vasodilators and vasoconstrictors (Fig. 12.1). Nitric oxide (NO) is an important and powerful vasodilator which is produced by the endothelium as a soluble gas with a short half-life (6–30 s in the artery wall and a few seconds in the blood). Its production involves a two-step oxidation of l-arginine to l-citrulline, with concomitant production of NO. This reaction is catalysed by NO-synthases (NOS) with the aid of cofactors, including tetrahydrobiopterin and nicotinamide adenine dinucleotide phosphate (NADPH) (Fig. 12.1). The NOS family plays a central role in the production of NO and consists of three different isoforms named after the tissues in which they were first identified: the neuronal (nNOS), inducible (iNOS) and endothelial (eNOS) isoforms. Many tissues can express more than one isoform, and all three may be constitutive or inducible [10]. Nonetheless, eNOS is the predominant form in endothelial cells and is the main source of endothelium-derived NO. eNOS is constitutively expressed and continuously produces small amounts of NO. eNOS can be stimulated by various hormones as well as haemodynamic factors. These stimuli induce an increase in intercellular Ca2+ that displaces the inhibitor caveolin from calmodulin to activate eNOS.
After production, NO diffuses to vascular smooth muscle cells (VSMCs) and activates guanylate cyclase, causing an increase in intracellular cyclic guanosine monophosphate (cGMP). This action leads to relaxation of the VSMCs and subsequent vasodilation. The importance of NO is supported by experimental work where inactivation of eNOS results in vasoconstriction and elevation in arterial blood pressure [11]. NO also exerts inhibitory effects on platelet aggregation, leukocyte adhesion and VSMC migration and proliferation, highlighting the importance of this hormone for vascular homeostasis [10].
12.2.1.2 Prostacyclin
The endothelium produces a family of prostaglandins through the catabolism of arachidonic acid by cyclooxygenases in response to mechanical and humoral stimuli. Prostacyclin (PGI2) is a major member of this family and acts as a paracrine-signalling molecule and activates the prostacyclin receptors on VSMC and platelets. This stimulates adenylate cyclase and with a consequent increase in intracellular levels of cyclic adenosine monophosphate (cAMP), ultimately resulting in VSMC relaxation and inhibition of platelet activation [12]. The action of PGI2 is closely related to that of NO since PGI2 potentiates NO release (and vice versa). Nonetheless, PGI2 seems less important for vascular control than NO, but plays a central role in the coagulation pathway (see Sect. 12.2.2).
12.2.1.3 Endothelium-Derived Hyperpolarizing Factor
Some of the endothelium-dependent vasodilation has generally been associated with hyperpolarization of the VSMCs and referred to a non-characterised factor called endothelium-dependent hyperpolarizing factor (EDHF) [13]. A number of candidate EDHFs have been suggested, including arachidonic acid metabolites, gaseous mediators (e.g. NO, hydrogen sulfide and carbon monoxide), reactive oxygen species, vasoactive peptides, potassium ions and adenosine. Irrespective of its exact nature, EDHF plays an important role in regulating vascular tone.
12.2.1.4 Endothelin
Endothelins are a family of potent vasoconstrictor peptides. Endothelin-1 (ET-1) is the predominant isoform and is primarily secreted by the endothelium in response to a variety of humoral and physical stimuli. After the production of ET-1 by the endothelium, it binds to the ETA and ETB receptors on VSMCs resulting in a sustained vasoconstriction. Endothelial cells also express ETB receptors whose stimulation results in the release of NO and PGI2, leading to vasodilation (and therefore serves as a feedback mechanism to partially oppose the vasoconstrictive effects of VSMC-located ETA/B-receptors). ET-1 also induces VSMC proliferation and growth in a dose-dependent manner, suggesting an important role for ET-1 to contribute to the atherosclerotic process [14].
12.2.1.5 Angiotensin
After cleavage of angiotensinogen to angiotensin (Ang) I via renin, this peptide is cleaved by the angiotensin-converting enzyme (produced by pulmonary and systemic vascular endothelium) into Ang II. The smooth muscle cell-localised AT1 receptor subtype mediates the predominant action of Ang II: vasoconstriction. These vasoactive actions are partly counteracted by the AT2 receptor, which causes vasodilatation [15]. Besides the vasoactive effects, Ang II leads to proliferation and growth of the VSMCs through activation of the AT1 receptor. Similarly to ET-1, the vasoconstrictive effects of AT1 are, at least partly, counterbalanced by a negative feedback loop in the vascular wall.
12.2.1.6 Thromboxane A2
Thromboxane A2 (TXA2) is an end product of arachidonic acid metabolism and is produced by TXA2 synthase. TXA2 is primarily produced by platelets, but also by the endothelium. The primary physiological role of TXA2 is platelet aggregation, but it has also been demonstrated to contribute to vasoconstriction [16].
12.2.1.7 Prostaglandin H2
In contrast to most members of the prostaglandin family, prostaglandin H2 (PGH2) is a vasoconstrictor substance. PGH2 is closely related to TXA2 as both are formed during arachidonic acid metabolism. Furthermore, PGH2 is the precursor of TXA2 and exerts its vascular effects through the same receptors [17].
12.2.2 Control of Blood Fluidity and Coagulation
The endothelium actively maintains an anticoagulant and antithrombotic surface through several mechanisms (Fig. 12.1). First, the endothelium keeps circulating platelets in a quiescent state, mainly through release of NO and PGI2 which synergistically increase cAMP content in platelets to repress activation and aggregation. Endothelial expression of ectonucleotidases also contributes to this process by converting ADP (a powerful trigger of platelet activation) to AMP and then adenosine. If platelet aggregation occurs, the release of serotonin and ADP from aggregating platelets will stimulate NO and PGI2 production to inhibit platelet aggregation and limit thrombus formation. Furthermore, vasodilation in response to NO and PGI2 serve to mechanically impede the progression of the coagulation process [18].
Secondly, endothelial cells promote the activity of anticoagulant pathways (Fig. 12.2). Anticoagulation is achieved through expression of thrombomodulin which interacts with thrombin, forming a complex that prevents activation of platelets or the conversion of fibrinogen to fibrin, a key step in the coagulation cascade. This complex also activates protein C, which works in combination with protein S to inactivate two essential cofactors for blood coagulation, VIIIa and Va. The endothelial surface layer (glycocalyx) contains heparan sulfate proteoglycans which binds and activates anti-thrombin III to inactivate thrombin and factors IXa, Xa and XIa. Finally, endothelial cells regulate initiation of coagulation by inhibiting the activation of factor X [19]. In addition to the strong anticoagulation effects, the endothelium can also contribute to coagulation. Endothelial expression of tissue factor (TF) enhances the activity of factor VII, which ultimately activates thrombin to facilitate activation of platelets and release of the von Willebrand factor (vWF) to further promote platelet aggregation.
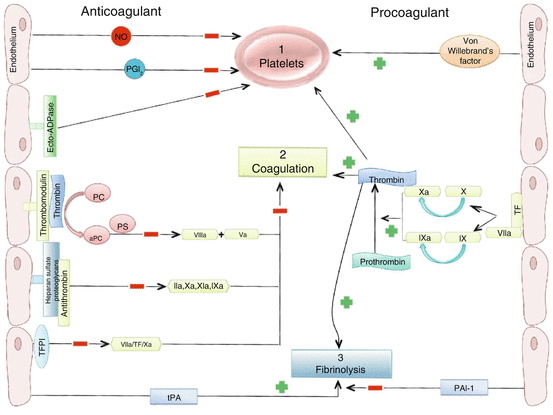
Fig. 12.2
Anticoagulant and procoagulant properties of the endothelium. The endothelium maintains blood fluidity through a balance between factors that either inhibit (left) or promote (right) (1) platelet activation, aggregation, and (2) coagulation, and inhibit (left) or promote (right) (3) fibrinolysis. NO nitric oxide, PGI 2 prostacyclin, PC protein C, PS protein S, TF tissue factor, TFPI tissue factor pathway inhibitor, tPA tissue-type plasminogen activator, PAI-1 plasminogen activator inhibitor 1
A third step in the coagulation pathway is the ability of the endothelium to influence fibrinolysis (Fig. 12.2) by the production of tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (uPA). These factors activate the liver-derived plasminogen into plasmin which then degrades fibrin. It is important to note that this activity is inhibited through the (endothelial) production of plasminogen activator inhibitor (PAI)-1 [19].
12.2.3 Inflammatory Responses
Under noninflammatory conditions, interaction of endothelial cells with leukocytes is suppressed by inhibiting the endothelium-dependent production of adhesion molecules (Fig. 12.1). Also NO production inhibits the fusion of Weibel-Palade bodies with the surface of the endothelial cell and leukocyte activation. However, during inflammation, a rapid response to infectious microbes or injured tissues occurs, involving local recruitment and activation of leukocytes. The purpose of the inflammatory response is to kill microbes and remove cellular debris. Endothelial cell activation in response to inflammation can be divided into rapid responses (type I) and slower responses (type II).
Type I responses are rapid (<10–20 min), transient and independent of protein synthesis. These responses generally initiate a signalling cascade that increases intracellular Ca2+ levels to serve a number of purposes. First, this response facilitates increased NO and PGI2 production, contributing to an increased blood flow and delivery of leukocytes. Secondly, increased Ca2+ levels enhance survival and migration of invading leukocytes and cause contraction of endothelial cells, which opens gaps between adjacent endothelial cells and increase permeability for leukocytes. Finally, expression of P-selectin and platelet-activating factor (PAF) is initiated which promotes the binding and activation of leukocytes [20].
Type II activation of endothelial cells involves a more persistent form of activation. During sustained inflammation, leukocytes produce inflammatory cytokines (e.g. tumour necrosis factor-α (TNF-α) and interleukin-1 (IL-1)) which results in the increased transcription of genes responsible for expression of a pro-adhesive and prothrombotic endothelial cell phenotype (IL-8 and adhesion molecules). Increased expression of these factors contributes to further leukocyte migration, adhesion and extravasation into the inflamed tissue. Inflammatory cytokines also induce leakage of plasma proteins into the affected tissue. Since these responses require transcription and translation of new proteins, type II activation is slower in onset but has more sustained effects than type I activation (i.e. hours–days). Accordingly, endothelial cells contribute to restoration of normal tissue architecture or form a connective tissue scar in response to inflammation [21].
12.2.4 Facilitation of Remodelling, Growth and Repair
Remodelling or adaptation of the vasculature refers to a basic compensatory response intended to maintain the functional integrity of the vessel in the presence of (potentially harmful) haemodynamic, metabolic and inflammatory stimuli. Sustained exposure to such stimuli, especially in conjunction with CVD risk factors, eventually transforms the initial (protective) response into a self-perpetuating and pathogenic process that contributes to the development of atherosclerosis (Fig. 12.3) [22].
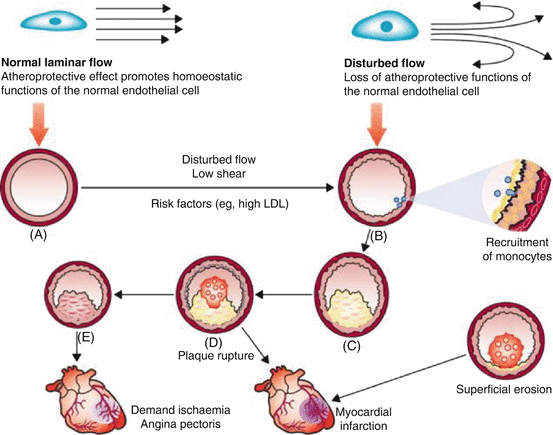
Fig. 12.3
Arterial remodelling influences the clinical consequences of atherosclerosis. Normal laminar shear stress maintains normal arterial calibre and properties (A). Disturbed flow characterised by a non-laminar, oscillatory flow promotes the upregulation of proatherogenic genes and recruitment of monocytes, as depicted to the right in the enlarged nascent plaque (B). Monocyte/macrophage accumulation yields a thin-capped, lipid-rich inflamed plaque (C), which can rupture and cause a thrombus (D), leading to myocardial infarction (central bottom). Alternatively, the plaque in (E) can undergo constrictive remodelling to promote flow-limiting stenosis that can cause demand ischaemia and angina pectoris. Less commonly, superficial erosion (bottom right) can cause myocardial infarction (With permission from Heusch et al. [22])
12.2.4.1 Remodelling
Straight portions of arteries are associated with laminar shear, which produces an atheroprotective genotype that mitigates the effects of risk factors. However, at flow dividers, disturbed flow impedes such atheroprotective functions and initiates increased expression of proatherogenic genes/proteins and (chronic) inflammatory responses. As a lesion forms and grows, matrix remodelling takes place, paving the way for abluminal expansion or outward growth of the growing atheroma that preserves the lumen of the artery and maintains blood flow. Ultimately, plaque growth can outstrip this compensatory enlargement of the artery wall, allowing the atheroma to encroach on the lumen and cause stenosis (Fig. 12.3). Smaller arterioles resist the atherosclerotic lesion formation. However, due to increases in pressure, these smaller vessels develop medial hypertrophy and intimal thickening, which sustains and aggravates hypertension [22].
Endothelial cells play a pivotal role in the process of remodelling. For example, early animal studies found that the presence of the endothelium is essential for arteries to adapt in luminal size [23]. When the endothelium is removed from an artery, exposure to elevations in shear stress does not induce a change in diameter. The adaptive response is therefore dependent on the endothelium, and more specifically, dependent on gene expression of eNOS and suppression of the bioavailability of ET-1 [24]. Also, immediate changes in vessel diameter in response to elevations in shear stress are dependent on an intact endothelium that releases vasoactive substances [25], highlighting the importance of the endothelium to respond (acutely and chronically) to changes in shear.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


