In translational models of pulmonary arterial hypertension (PAH), spironolactone improves cardiopulmonary hemodynamics by attenuating the adverse effects of hyperaldosteronism on endothelin type-B receptor function in pulmonary endothelial cells. This observation suggests that coupling spironolactone with inhibition of endothelin type-A receptor–mediated pulmonary vasoconstriction may be a useful treatment strategy for patients with PAH. We examined clinical data from patients randomized to placebo or the selective endothelin type-A receptor antagonist ambrisentan (10 mg/day) and in whom spironolactone use was reported during ARIES-1 and -2, which were randomized, double-blind, placebo-controlled trials assessing the effect of ambrisentan for 12 weeks on clinical outcome in PAH. From patients randomized to placebo (n = 132) or ambrisentan (n = 67), we identified concurrent spironolactone use in 21 (15.9%) and 10 (14.9%) patients, respectively. Compared with patients treated with ambrisentan alone (n = 57), therapy with ambrisentan + spironolactone improved change in 6-minute walk distance by 94% at week 12 (mean ± SE, +38.2 ± 8.1 vs +74.2 ± 27.4 m, p = 0.11), improved plasma B-type natriuretic peptide concentration by 1.7-fold (p = 0.08), and resulted in a 90% relative increase in the number of patients improving ≥1 World Health Organization functional class (p = 0.08). Progressive illness, PAH-associated hospitalizations, or death occurred as an end point for 5.3% of ambrisentan-treated patients; however, no patient treated with ambrisentan + spironolactone reached any of these end points. In conclusion, these pilot data suggest that coupling spironolactone and endothelin type-A receptor antagonism may be clinically beneficial in PAH. Prospective clinical trials are required to further characterize our findings.
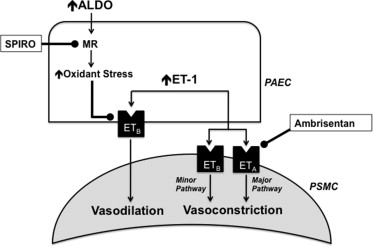
The potential contribution of aldosterone to the pathobiology of pulmonary artery hypertension (PAH) has been suggested by several groups, although the clinical consequences of aldosterone inhibition with spironolactone in patients with PAH remain unresolved. We have demonstrated recently that aldosterone levels are increased significantly in the pulmonary arterial circulation of patients with PAH; and in experimental animal models of PAH, this modulates pulmonary vascular dysfunction by inhibiting endothelin type-B receptor–dependent synthesis of nitric oxide. This observation suggests that coupling therapies that inhibit endothelin type-A receptor–mediated pulmonary vasoconstriction with spironolactone may be a useful strategy to treat PAH ( Figure 1 ). To test this hypothesis, we retrospectively analyzed the Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Study 1 (ARIES-1) and Study 2 (ARIES-2) to determine if treatment with spironolactone influenced the efficacy of the selective endothelin type-A receptor antagonist ambrisentan on the predetermined study end points.
Methods
Patients with World Health Organization (WHO) group I PAH who had received treatment in the randomized, placebo-controlled phase III trials ARIES-1 (n = 201) and ARIES-2 (n = 192) were eligible for analysis. The full methods and results for both studies have been reported previously. Patients received for 12 weeks either placebo or the selective endothelin type-A receptor antagonist ambrisentan at 5 or 10 mg/day orally in ARIES-1 or 2.5 or 5 mg/day orally in ARIES-2. The primary end point for both studies was the change from baseline in the 6-minute walk distance (6-MWD) at week 12. The secondary end points included in the study that were relevant to the current analysis included plasma B-type natriuretic peptide (BNP) concentration, change in WHO functional class, and total clinical worsening events, which were a composite of PAH-associated hospital admissions, early study termination due to rapidly progressive illness (see the study by Galiè et al for early escape criteria) and death.
To determine retrospectively the effect of ambrisentan + spironolactone on outcome in PAH, we accessed the ARIES-1 and ARIES-2 database to identify patients randomized to either placebo or ambrisentan and reported concurrent spironolactone use at any dose. Consideration of patients for inclusion in the spironolactone treatment groups in the current analyses was based on the following criteria achieved during the course of the original ARIES trial: (1) the duration of spironolactone use was for ≥28 days, (2) spironolactone therapy was initiated before study enrollment or ≤28 days after the first dose of ambrisentan/placebo, and (3) discontinuation of spironolactone did not occur within 28 days before the final ambrisentan/placebo dose. We elected to study patients receiving the maximum trial dose of ambrisentan (10 mg/day, n = 67) to test our hypothesis, because it was at this treatment dose that ambrisentan induced the strongest clinical and hemodynamic effects in the ambrisentan development program.
The demographic and baseline characteristics were summarized by treatment group (placebo, spironolactone, ambrisentan [10 mg/day, without concurrent spironolactone use], ambrisentan [10 mg/day] + spironolactone) and differences among treatment groups were analyzed using the Student t test for most continuous variables. The Wilcoxon rank sum test was used to analyze differences among treatment groups for WHO functional class as well as study drug and spironolactone dosing comparisons, and Fisher’s exact test was used for categorical variables. Plasma BNP concentrations were summarized with logarithmic transformation. The change from baseline in WHO functional class is presented categorically and was analyzed with a 7-point scale: −3 to −1 is improved, 0 is no change, and +1 to +3 is deterioration as reported previously. The last observation carried forward approach was used for 6-MWD and WHO functional class. Only the patients with both a baseline and week-12 value were included in the BNP geometric mean ratio (GMR) analysis, which is a comparison of the geometric mean BNP at week 12 relative to the geometric mean BNP at baseline. To determine if accounting for spironolactone use in this study influenced the effect of ambrisentan on the primary and secondary end points as observed in the original ARIES trial, differences between placebo versus ambrisentan or ambrisentan + spironolactone were assessed before analyzing differences between the ambrisentan and ambrisentan + spironolactone treatment groups. For all analyses, p <0.05 was considered significant. No adjustments were made for multiple comparisons.
Results
Of patients randomized to placebo (n = 132) or ambrisentan 10 mg/day (n = 67), concurrent spironolactone use was identified for 21 (15.9%) and 10 (14.9%) patients, respectively. Compared with ambrisentan alone (n = 57), patients in the ambrisentan + spironolactone group (n = 10) were more likely to have WHO functional class III/IV and tended to have worse baseline 6-MWD, pulmonary vascular resistance, and plasma BNP concentration, although no clinically meaningful differences between treatment groups were observed with respect to patient demographics, Borg Dyspnea index, cardiac index, or right atrial pressure ( Table 1 ).
| Variable | Placebo | Ambrisentan (10 mg/day) | p | ||
|---|---|---|---|---|---|
| −SPIRO (n = 111) | +SPIRO (n = 21) | −SPIRO (n = 57) | +SPIRO (n = 10) | ||
| Age (yrs) | 50 (18–81) | 47 (27–69) | 50 (18–78) | 47 (24–78) | 0.58 |
| Women | 87 (78.4%) | 16 (76.2%) | 45 (78.9%) | 8 (80.0%) | 1.00 |
| Race | |||||
| Caucasian | 84 (75.7%) | 16 (76.2%) | 35 (61.4%) | 9 (90.0%) | 0.15 |
| Black | 4 (3.6%) | 0 | 2 (3.5%) | 1 (10.0%) | |
| Asian | 4 (3.6%) | 0 | 1 (1.8%) | 0 | |
| Hispanic | 19 (17.1%) | 5 (23.8%) | 17 (29.8%) | 0 | |
| Other | 0 | 0 | 2 (3.5%) | 0 | |
| Pulmonary arterial hypertension origin | |||||
| Primary pulmonary hypertension | 66 (59.5%) | 19 (90.5%) | 34 (59.6%) | 7 (70%) | 0.73 |
| Nonprimary pulmonary hypertension (CTD; HIV; Anorexigen) | 45 (40.5%) | 2 (9.5%) | 23 (40.4%) | 3 (30%) | |
| Baseline WHO functional class | |||||
| I | 4 (3.6%) | 0 | 2 (3.5%) | 0 | 0.31 |
| II | 40 (36.0%) | 7 (33.3%) | 20 (35.1%) | 2 (20%) | |
| III | 55 (58.6%) | 13 (61.9%) | 31 (54.4%) | 5 (50%) | |
| IV | 2 (1.8%) | 1 (4.8%) | 4 (7.0%) | 3 (30%) | |
| Borg dyspnea index | 3.8 ± 2.1 | 4.3 ± 2.4 | 3.8 ± 2.1 | 3.8 ± 2.1 | 0.95 |
| 6-MWD (m) | 346.7 ± 75.3 | 318.9 ± 97.7 | 344.8 ± 79.0 | 322.3 ± 74.9 | 0.41 |
| Cardiac index (L/min/m 2 ) | 2.4 ± 0.8 | 2.4 ± 0.8 | 2.6 ± 0.7 | 2.4 ± 0.8 | 0.44 |
| Pulmonary vascular resistance (Wood units) | 11.3 ± 6.8 | 12.6 ± 7.2 | 10.8 ± 5.7 | 14.5 ± 6.0 | 0.07 |
| Mean pulmonary artery pressure (mm Hg) | 49.7 ± 14.2 | 57.0 ± 12.7 | 50.4 ± 15.5 | 57.4 ± 19.0 | 0.21 |
| Mean right atrial pressure (mm Hg) | 7.5 ± 4.8 | 9.4 ± 5.9 | 8.8 ± 5.0 | 11.2 ± 8.7 | 0.22 |
| Plasma B-type natriuretic peptide (ng/L) | 130.8 (99.1–172.7) | 142.4 (78.9–257.2) | 131.7 (88.0–197.2) | 236.7 (81.5–687.4) | 0.24 |
The dose, duration, and clinical indication for spironolactone therapy are listed in Tables 2 and 3 . Spironolactone therapy was initiated after the start of the ARIES trial date for 2 patients in the ambrisentan + spironolactone group (day 15 and 21) and for 2 patients in the spironolactone group (day 9 and 29), and the start date of spironolactone therapy was before ARIES but unknown for 2 patients in the spironolactone group. Spironolactone therapy initiation for the remaining patients in the spironolactone (n = 17) and ambrisentan + spironolactone (n = 8) groups occurred well in advance of patients’ first ARIES trial date and did not differ significantly between groups. In both placebo (n = 21) and ambrisentan (n = 10) conditions, spironolactone therapy was reported for ≥96% of ARIES drug days, and no significant difference was observed for mean ± SD daily spironolactone dose between groups. Of patients in the ambrisentan-only condition (n = 57), 6 patients (10.5%) identified as spironolactone users did not meet the criteria for inclusion in the ambrisentan + spironolactone condition. For these patients, the first dose of spironolactone therapy occurred at a mean ± SD of 38 ± 13 days after ARIES drug initiation, and the mean ± SD duration of spironolactone use in these patients was 21 ± 22 days, which spanned 29 ± 24% of the ARIES drug days.
| Treatment Characteristic | Spironolactone Treatment Condition | p | |
|---|---|---|---|
| Placebo, n = 21 (SD) | Ambrisentan, n = 10 (SD) | ||
| Number of days on SPIRO therapy before ARIES drug initiation | 294 (352) ∗ | 280 (312) † | 0.68 |
| Number of ARIES drug days | 80 (15) | 85 (3) | 0.93 |
| Number of days on study drug in ARIES in which SPIRO was prescribed | 78 (16) | 81 (8) | 0.93 |
| % ARIES days that patients were prescribed SPIRO | 97 (8) | 96 (9) | 0.64 |
| Daily SPIRO dose (mg/day) | 39 (15) | 31 (12) | 0.17 |
| Clinical Indication for Spironolactone ∗ | Spironolactone Treatment Condition | |
|---|---|---|
| Placebo (n = 21) | Ambrisentan (n = 10) | |
| Pulmonary hypertension | 14 | 7 |
| Edema or prevention of edema | 6 | 5 |
| RV failure | 4 | 3 |
| Electrolyte imbalance | 1 | 0 |
We first analyzed the change in 6-MWD from baseline between placebo and ambrisentan-treated patients. We observed that compared with placebo, ambrisentan improved 6-MWD at week 4 (mean ± SE: +17.5 ± 5.6 m), week 8 (+32.8 ± 7.1 m), and week 12 (+38.2 ± 8.1 m), whereas a −12.1 ± 8.5 m decrease in 6-MWD was observed at week 12 for placebo-treated patients (p <0.01 at week 12). Similarly, we observed that compared with placebo, patients treated with ambrisentan + spironolactone demonstrated a significantly improved change in 6-MWD from baseline at week 4 (mean ± SE: +59.9 ± 22.2 m), week 8 (+66.3 ± 31.9 m), and week 12 (+74.2 ± 27.4 m, p <0.01). Collectively, these trends agree with findings reported in the ARIES-1 and ARIES-2 trials, and indicate that the effect of ambrisentan on the predetermined primary end point was not contingent on concurrent treatment with spironolactone. We next analyzed the effect of spironolactone on change in 6-MWD from baseline in ambrisentan-treated patients. We observed that ambrisentan + spironolactone therapy patients outperformed ambrisentan-treated patients at each measured time point, ultimately achieving a peak change in 6-MWD distance at week 12 (mean ± SE, +59.9 ± 22.2 vs +17.5 ± 5.6 m [week 4], +66.3 ± 31.9 vs +32.8 ± 7.1 m [week 8], +74.2 ± 27.4 vs +38.2 ± 8.1 m [week 12], p = 0.11 at week 12; Figure 2 ), which was associated with a +13.6 m difference in the absolute 6-MWD at week 12 (383.0 ± 12.3 vs 396.6 ± 26.3 m).



