Fig. 4.1
Cardiac Sarcomere. The sarcomere is the basic contractile unit of muscle. The lateral boundaries of a sarcomere are defined by protein-dense Z-discs. The I-band is the region surrounding the Z-disc in which actin-containing thin filaments do not overlap with the myosin-containing thick filaments. The A-band comprises the region extending the entire length of the thick filaments, while the H-zone is the region in the center of the sarcomere, devoid of thin filaments. The enlarged region depicts the actin-thin filament. CapZ caps the barbed end of the thin filament, while Tmod1 caps the pointed end. Lmod2 binds to the thin filament pointed end and likely competes with Tmod1 to fine-tune thin filament length. Cap2 binds in the region of the M-band in adult myofibrils. One tropomyosin (orange) associates with every seven actins and a troponin complex. Nebulette localizes to the Z-disc
In order for actin to function efficiently in muscle contraction, it must be organized within the sarcomere to the level of single molecules. The sarcomere is composed of numerous structural and regulatory proteins, with the three prominent cardiac filaments consisting primarily of actin (thin filaments), myosin (thick filaments), and titin. The lateral boundaries of the sarcomere are defined by protein-dense structures known as Z-discs. Actin-thin filaments are precisely organized in striated muscle with the filament barbed ends inserted in the Z-disc and their pointed ends extending out to the center of the sarcomere (M-line) where they overlap with myosin (motor protein)-containing thick filaments. The terminology of barbed and pointed end came from the arrowhead morphology of myosin S1 fragments bound to actin filaments in vitro. Titin is the largest protein known in humans. One titin molecule spans half of a sarcomere and functions as a molecular spring that provides the passive tension in muscle.
The numerous contractile and regulatory proteins that constitute the sarcomere are precisely orchestrated to generate the force necessary for muscle contraction. In fact, approximately 60 years ago the sliding filament theory of muscle contraction was first proposed (Huxley and Niedergerke 1954; Huxley and Hanson 1954; Huxley 1985), which states that myosin remains stationary in the sarcomere and during contraction the thin filament slides past myosin to generate tension. Remarkably, Huxley’s sliding filament theory was proposed prior to the discovery of most of the regulatory molecules involved in muscle contraction, and while it has been refined over the years, this model is the basis for current models of muscle contraction (reviewed in Cooke 2004; Szent-Györgyi 2004; Williams 2011).
4.2 Major Components of the Thin Filament
The thin filament, while composed of a core of filamentous actin (F-actin), also contains many regulatory proteins that are necessary for efficient contraction. Sarcomeric F-actin polymerizes into a helical filament with one tropomyosin coil–coil associating with every seven actin molecules. Associated with each tropomyosin coil–coil is a troponin (Tn) complex, which consists of TnI (inhibitory), TnT (tropomyosin-binding), and TnC (calcium-binding) (Fig. 4.1). The regulation of contraction is controlled by calcium and the tropomyosin/troponin complexes (reviewed in Kobayashi and Solaro 2005). These regulatory thin filament proteins are also hotspots for numerous mutations that have been linked to both dilated and hypertrophic cardiomyopathies (for review, see Tardiff 2011).
4.3 Thin Filament Length Regulation
Strict regulation of uniform thin filament lengths in striated muscle is necessary for proper muscle contraction. Interestingly, thin filament lengths vary based on species (Burkholder and Lieber 2001) and even between different types of muscles (Granzier et al. 1991; Kruger et al. 1991). In addition, thin filament lengths can vary between different regions of the heart. For instance, the stiffer papillary muscle has a narrower range of thin filament lengths when compared to the less stiff atria muscles (Robinson and Winegrad 1977), indicating thin filament lengths may be fine-tuned based on the requirements (e.g., load) of specific muscle types. In cardiac muscle, thin filament lengths have a broader range of lengths when compared to skeletal muscle (Page and Huxley 1963; Robinson and Winegrad 1977; Littlefield and Fowler 2002; Burgoyne et al. 2008). The implications of a broader range of lengths in cardiac muscle is an interesting, yet unexplored, field of research.
Thin filaments are not static structures. Uniform thin filament lengths are maintained even with continuous dynamic exchange of actin monomers at the ends of the filament. Based on the 3–10 day half-life of the major components of the thin filament (actin, troponin complex, and tropomyosin) (Martin 1981), it has been estimated that actin monomers can move in and out of the cardiac thin filament an average of over 100 times in their lifetime (reviewed in Littlefield and Fowler 1998). A class of molecules known as capping proteins contributes to the maintenance of thin filament lengths. In vitro experiments have demonstrated that actin monomers are strongly inhibited from exchanging into the thin filament when the filaments ends are bound by capping proteins. The two primary striated muscle capping proteins that have been identified are tropomodulin 1 (Tmod1) and CapZ, which bind to the pointed and barbed end of the thin filament, respectively (Casella et al. 1986; Weber et al. 1994). Remarkably, although in live cardiomyocytes, CapZ and Tmod1 are transiently bound to the thin filament, allowing monomer incorporation at both pointed and barbed ends, the lengths of the filaments are not altered (Littlefield et al. 2001). Therefore, the dynamics of capping proteins are critical in determining the length of thin filaments.
4.4 Capping Proteins: Tmod and CapZ
Tmod1 plays a key role in regulating and stabilizing thin filament pointed ends by blocking monomeric, globular (G-actin) actin incorporation and dissociation in vitro (Weber et al. 1994). The Tmod family of proteins contain four isoforms encoded by separate genes (Tmod1-4), with Tmod1 being the primary isoform in the heart (Fowler 1996). Tmod1 is expressed in multiple tissues and was originally described in red blood cells as a tropomyosin-binding partner (Fowler 1987). In striated muscle, Tmod1 binds to thin filament pointed ends via its two actin-binding domains and two tropomyosin-binding domains (Babcock and Fowler 1994; Weber et al. 1994; Kostyukova et al. 2000, 2001, 2006; Fowler et al. 2003). Tmod1 is necessary for embryonic development, demonstrated by genetically engineered Tmod1 null mice dying at E10.5; at death the embryos displayed numerous defects including failure of the heart to loop and aborted cardiac myofibrillogenesis (Chu et al. 2003; Fritz-Six et al. 2003). The heart was the primary defect in the Tmod1 deficient mice as cardiac-specific expression of Tmod1 rescued the embryonic lethality (McKeown et al. 2008).
Further insights into the role of Tmod1 in cardiac muscle were demonstrated by experiments that showed that when barbed-end incorporation of fluorescent rhodamine-actin was blocked (via CapZ), thin filament lengths were not changed while overexpression of GFP-tagged Tmod1 resulted in shorter thin filament lengths (Littlefield et al. 2001). Additionally, in isolated live cardiomyocytes, a higher ratio of rhodamine-actin incorporated into the pointed end compared to the barbed end. Together these results suggest that the pointed end was more dynamic than the barbed end. The consequences of altering thin filament lengths in vivo are not well studied and is an exciting area of active research.
As described above, thin filament lengths are highly dependent on the cellular levels of Tmod1. Other examples include, in neonatal rat cardiomyocytes, overexpression of Tmod1 resulted in shorter actin filament lengths and myofibril degeneration, while knockdown of Tmod1 resulted in longer actin filaments (Sussman et al. 1998a). Altering the levels of Tmod1 might change the exchange and/or dynamics of Tmod1 at the pointed end. For example, an increase in Tmod1 protein expression may change it from a dynamic to a stable cap, which could potentially explain the observed reduction in thin filament lengths upon overexpression. Additionally, when the interaction of Tmod1 with tropomyosin was blocked in isolated chick cardiomyocytes (via an anti-Tmod1 antibody), actin filament elongation occurred and cell beating was compromised (Gregorio et al. 1995), indicating loss of thin filament length control had a detrimental effect on contractile function.
Within the Tmod family are the leiomodin (Lmod) proteins. While the Tmod family has been studied for over 25 years (Fowler 1987), the Lmod proteins were identified over a decade later (Conley et al. 2001). Lmod has three isoforms encoded by separate genes: smooth muscle (Lmod1), cardiac/skeletal (Lmod2), and fetal/skeletal (Lmod3) (Conley et al. 2001). Lmod and Tmod proteins are approximately 40 % identical at the protein level (Chereau et al. 2008). Lmods differ from Tmods in that they do not contain a second tropomyosin-binding domain and have a C-terminal extension that, importantly, contains a third actin-binding site [Wiskott–Aldrich syndrome protein 2 (WH2) domain (reviewed in Yamashiro et al. 2012)]. Due to the presence of a third actin-binding site, Lmod2 has potent nucleation abilities in vitro (Chereau et al. 2008). Surprisingly, although similar in domain structure to Tmod1, currently there is no evidence that Lmod2 “caps” actin-thin filament pointed ends (Tsukada et al. 2010). However, when the unique C-terminal extension of Lmod2 is removed, actin filament capping activity is detected, indicating that the C-terminus of Lmod2 acts to blunt its ability to cap the pointed end (Tsukada et al. 2010). The functional properties of Lmod2 in vivo have not been reported.
While Lmod2 is the most well-studied Lmod isoform, little is known about the role of Lmod2 in actin filament assembly and regulation. However, like Tmod1, Lmod2 has been shown to bind to the pointed end of thin filaments in primary cardiomyocytes (Tsukada et al. 2010). Manipulation of Lmod2 levels also alters thin filament lengths. In fact, Lmod2 was the first mammalian pointed end elongation factor identified, as overexpression of Lmod2 displaced Tmod1 and resulted in longer thin filaments (Tsukada et al. 2010). In Drosophila, SALS (sarcomere length short), similar but not homologous to Lmod2, has a WH2-binding domain as well as a proline-rich domain and is localized to the pointed end. Like Lmod2, overexpression of SALS elongated thin filaments, while knockdown shortened thin filament lengths (Bai et al. 2007).
One current model is that Lmod2 and Tmod1 have an antagonistic relationship in which they compete for binding to the pointed end, thus contributing to the fine-tuning of thin filament lengths (Fig. 4.2). When Lmod2 is present at the pointed end, Tmod1 cannot bind and thin filaments elongate. Alternatively, when Tmod1 is present on the pointed end, Lmod2 cannot bind and lengths are reduced (Tsukada et al. 2010). Another model proposes that there are two populations of actin filaments: more stable core filaments associated with Tmod1 and more dynamic filaments associated with Lmod2 (Skwarek-Maruszewska et al. 2010; Tsukada et al. 2010). Further investigation is necessary to determine how Tmod and Lmod coordinate to regulate thin filament length.
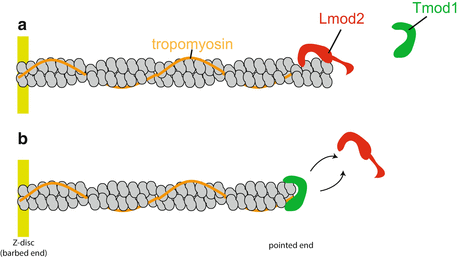
Fig. 4.2
Model for thin filament length regulation at the pointed end. Lmod2 and Tmod1 compete to bind to the thin filament pointed end and function to fine-tune thin filament length. (a) When Lmod2 is bound to the pointed end, Tmod1 cannot bind and the thin filament elongates. (b) Conversely, when Tmod1 binds to the pointed end, Lmod2 cannot bind and lengths are restricted. Lmod2 and Tmod1 exchange from the pointed end to maintain proper thin filament length. Figure modified from Tsukada et al. (2010)
Lmod2 binds to mature myofibrils and localization is dependent on the contractile state of the cardiomyocytes and a dynamic actin monomer population. Treatment of cardiomyocytes with blebbistatin (a myosin II inhibitor that reduces contractility) resulted in the loss of Lmod2 localization at the pointed end. Cardiomyocytes treated with Latrunculin B, which binds actin monomers and prevents their polymerization, also resulted in loss of pointed-end localization of Lmod2. Interestingly, Tmod1 localization was unaffected by blebbistatin and Latrunculin B treatment. These experiments suggest that the localization of Lmod2 (but not Tmod1) requires an actin monomer pool capable of polymerization (Skwarek-Maruszewska et al. 2010). Lmod2 likely plays an important role in the maintenance of thin filament lengths. However, functional studies of the physiological role of Lmod2 in vivo are necessary.
CapZ (also known as “capping protein”) binds to the barbed end of thin filaments in striated muscle Z-discs and stabilizes the filaments (Isenberg et al. 1980; Casella et al. 1987; Caldwell et al. 1989). Capping protein is a heterodimeric protein composed of α and β subunits (Isenberg et al. 1980). Vertebrates have three α subunits encoded by three different genes and three β subunits that result from alternate splicing of a single gene (Hurst et al. 1998; Hart et al. 2000). Capping protein isoforms display tissue-specific distributions (Yamashita et al. 2003). The isoform α1β1 is the primary isoform that localizes to the Z-disc and is referred to as CapZ (Casella et al. 1987). CapZ is a highly dynamic protein which binds transiently to the thin filament and regulates polymerization and depolymerization of thin filaments (Wear et al. 2003).
The C-terminal extension of each CapZ α and β subunit (tentacles) interacts with one terminal actin molecule (Fig. 4.3a) (Yamashita et al. 2003). Binding of the β-tentacle to actin is critical for capping, as mutations in the β-tentacle increased actin dynamics while mutations in the α-tentacle did not significantly affect actin dynamics (Barron-Casella et al. 1995; Wear et al. 2003; Kim et al. 2010; Lin et al. 2013). In order for actin exchange to occur, the CapZ α-tentacle dissociates, with only the β-tentacle still attached (Yamashita et al. 2003).
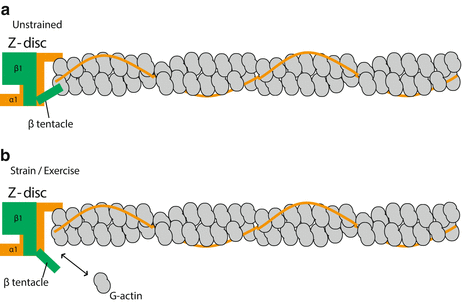
Fig. 4.3
Model of how CapZ interacts with actin at the Z-disc. (a) When unstrained, CapZ caps the barbed end of the thin filament. The CapZ α1 and β1 tentacles each bind to one actin, which decreases the off rate of actin (less dynamic). (b) Following cyclic strain (mimics exercise), CapZ β1 tentacle undergoes a putative structural change that results in increased actin dynamics. This figure was modified from Lin et al. (2013)
Modest CapZ reduction in transgenic mice resulted in blunted protein kinase C (PKC)-mediated myofilament inhibition (Pyle et al. 2002, 2006). PKC inhibition is protective against the development of hypertrophy and CapZ reduction resulted in increased baseline contractility and calcium sensitivity compared with wild-type mice (Gaikis et al. 2013), indicating CapZ may also be important in the hypertrophic response. In addition, modest CapZ reduction in mice is cardioprotective following ischemia-reperfusion injury (Yang and Pyle 2012). CapZ clearly has diverse biological functions, not only as a barbed-end capping protein but may also play a role in intracellular signaling and the cardiac hypertrophy response.
4.5 Stabilization of the Thin Filament
Tropomyosin is an α-helical protein that dimerizes in a head-to-tail fashion along every seven actin monomers and stabilizes the thin filament. Vertebrates have four tropomyosin genes (TPM1–TPM4) that generate numerous isoforms through alternative splicing: TPM1α is the predominant isoform in adult striated muscle (Schevzov et al. 2005; Dube et al. 2014). Tropomyosin is vital to cardiac function as mice deficient in α-tropomyosin are embryonic lethal at E9.5 (Blanchard et al. 1997). Thin filaments depolymerize more slowly when stabilized by tropomyosin (Broschat et al. 1989; Broschat 1990). Tropomyosin also stabilizes the thin filament by blocking depolymerization by the actin severing proteins cofilin (Nishida et al. 1984; Ono and Ono 2002) and gelsolin (Ishikawa et al. 1989; Khaitlina et al. 2013). In addition, the interaction of Tmod and tropomyosin enhances the efficiency of Tmod capping actin filaments in vitro (Weber et al. 1994, 1999; Gregorio and Fowler 1995). In other words, Tmod1 is a “leaky” cap in the absence of tropomyosin and can only partially inhibit polymerization and depolymerization (Weber et al. 1994, 1999; Kostyukova et al. 2005). In this regard, X-ray crystallography revealed a mechanism by which Tmod1 functions as a leaky cap. Specifically, one Tmod1 can bind to the thin filament pointed end through multiple distinct low affinity interactions with tropomyosin and actin; these weak interactions permit only part of Tmod1 to detach from the pointed end, allowing for actin exchange (Rao et al. 2014).
In skeletal muscle, the giant sarcomeric protein nebulin (~700–800 kDa) spans the length of the actin-thin filament, where it can interact with Tmod1 in the center of the sarcomere (McElhinny et al. 2001) and CapZ in the Z-disc (Wang and Wright 1988; Pappas et al. 2008). Nebulin knockout mice have shorter skeletal thin filament lengths and die within 2 weeks after birth (Bang et al. 2006; Witt et al. 2006; Gokhin et al. 2009). Nebulin was proposed to be a “molecular ruler” that specified thin filament lengths (for reviews, see McElhinny et al. 2005; Horowits 2006; Pappas et al. 2011). Notably, the molecular ruler hypothesis has been difficult to prove due to the large size of nebulin and its susceptibility to proteolysis. To directly test the ruler hypothesis, a shorter version of human nebulin called “mini-nebulin” was synthesized. Mini-nebulin contains all the unique C- and N-terminal regions of nebulin but has only 4 of nebulin’s 22 super repeats (Pappas et al. 2010). Introduction of mini-nebulin into cultured primary skeletal myocytes did not alter the lengths of the thin filaments. However, upon exposure to a depolymerization agent (Latrunculin A), thin filament lengths shortened to the length of mini-nebulin. This experiment supports the idea that nebulin plays an important role in stabilization of thin filaments and likely does not function as a strict ruler (Pappas et al. 2010). As such, current evidence suggests a two-segment model of thin filament length stabilization in skeletal muscle: (1) a core filament that is coated and stabilized by nebulin extending from the Z-disc to approximately 0.95 μm and (2) a nebulin-free region extending from 0.95 μm to the pointed end that is variable in length and regulated by Tmod1 via a nebulin-independent mechanism (reviewed in Gokhin and Fowler 2013).
While nebulin is highly abundant in skeletal muscle, it is a minor component in cardiac muscle (Kazmierski et al. 2003). Nebulin null mice present with no detectable cardiac dysfunction prior to premature death at 2 weeks old (Bang et al. 2006; Witt et al. 2006). Thus, the generation of a cardiac nebulin-specific knockout model in the future will assist in determining if nebulin has a role in thin filament length regulation in the heart.
A much smaller cardiac-specific nebulin-like protein, nebulette (encoded by a separate gene), localizes to the Z-disc (Moncman and Wang 1995; Millevoi et al. 1998). Nebulette is far less studied in comparison to nebulin. Nebulette also seems unlikely to act as a molecular ruler because it is only localized to the Z-disc, and it is too small to extend along the thin filament. Interestingly, however, knockdown of nebulette in cardiomyocytes resulted in shorter thin filament lengths (Moncman and Wang 2002), suggesting that the role of nebulette (like nebulin) in thin filament length regulation may include a stabilization mechanism. A nebulette knockout mouse has yet to be published.
4.6 Dynamic Equilibrium Between G-Actin and F-Actin
Another potential method of thin filament length control is the equilibrium between the pool of G-actin (globular actin) and F-actin (filamentous actin). Actively contracting sarcomeres have a high rate of actin turnover (Martin 1981). Therefore, proteins that bind or sequester G-actin likely contribute to thin filament length. Cyclase-associated proteins (CAPs), for example, are G-actin sequestering proteins. Vertebrates have two isoforms, CAP1 and CAP2. CAP1 is ubiquitously expressed and primarily functions in cell polarity and motility, while CAP2 is expressed in the brain, heart, skeletal muscle, and skin. CAP2 has a nuclear localization pattern during myofibrillogenesis, but is localized to the M-line (possibly binds to the actin thin filament pointed ends but this remains to be confirmed) in adult myofibrils (Peche et al. 2007). CAP2 is proposed to regulate thin filament stabilization and actin dynamics by severing F-actin and preventing polymerization by sequestering the G-actin pool (Fig. 4.4) (Peche et al. 2013). A direct link between CAP2 and filament length regulation has not yet been shown.
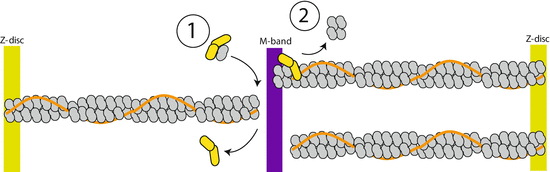
Fig. 4.4
Cap2 localizes to the region of M-band. The mechanisms by which Cap2 could maintain thin filament length is (1) to sequester and maintain a pool of G-actin for polymerization and/or (2) sever F-actin. Figure modified from Peche et al. (2013)
Actin-monomer-binding proteins likely work in conjunction with capping proteins to regulate thin filament length. In Caenorhabditis elegans striated muscle, Tmod1 has been shown to act synergistically with profilin (binds G-actin) and ADF/cofilin (severs F-actin) to prevent elongation of thin filaments. The dynamic exchange of Tmod1 allows free pointed ends to elongate; loss of Tmod1 at the pointed end leads to a transient tropomyosin-free pointed end. Data suggests that ADF/cofilin binds to the tropomyosin-free pointed end and severs the filament. Profilin can then bind to the depolymerized G-actin to prevent elongation of the thin filament (Yamashiro et al. 2008). While thin filament length could not be directly measured (due to the severe sarcomeric disorganization observed), it is postulated that the cooperation between Tmod1 capping with profilin and cofilin could be a mechanism to regulate thin filament length due to the clear role Tmod1 plays in length regulation. The potential role of proteins that bind or sever actin in thin filament length regulation has yet to be directly tested, but likely are important players in length regulation.
4.7 Links Between Alternations in Thin Filament Components and Myopathies
A notable link between thin filament length regulation in the heart and disease is that Tmod1 overexpression transgenic mice (TOT) develop dilated cardiomyopathy (DCM). Notably, TOT mice display shorter I-band length, which suggests shorter thin filament lengths [as was directly measured in isolated primary myocytes overexpressing Tmod1 (Sussman et al. 1998a, b)]. Although the Lmod2 gene is hypothesized to be involved in hypertrophic cardiomyopathy (HCM), due to its location near the HCM locus CMH6 (Conley et al. 2001), no studies to date have linked Lmod2 to the progression of cardiomyopathy.
Myopathies related to thin filament length have also been noted in skeletal muscle. Mutations in the giant sarcomeric protein nebulin result in nemaline myopathy, and the muscle weakness associated with this disease has been linked to shorter thin filament lengths (Ottenheijm et al. 2010, 2013; Lawlor et al. 2011). The effect of thin filament length on contractile function has been studied in skeletal muscle (Granzier et al. 1991; Bang et al. 2006; Witt et al. 2006; Chandra et al. 2009; Gokhin et al. 2009; Ottenheijm et al. 2009), but it is not as well defined for cardiac muscle. For instance, nebulin knockout mice with shorter skeletal muscle thin filament lengths have reduced contractile function but the effect of nebulin knockout on heart contractile activity is not known (Bang et al. 2006; Witt et al. 2006; Gokhin et al. 2009).
Alteration in expression and localization of CapZ results in HCM. Cardiac-specific overexpression of the CapZ β2 isoform (which normally localizes to the intercalated disc) in mice inhibits expression and localization of the CapZ β1 isoform (Z-disc) and results in the development of HCM (Hart and Cooper 1999). From this study it was suggested that an important function of CapZ is to bind to the Z-disc and anchor actin filaments; loss of CapZ localization leads to myofibrillar disarray and HCM. Further evidence supporting the role of CapZ in HCM came from BAG3 (bcl-2-associated athanogene) knockout mice which show mislocalization of CapZ from the Z-disc resulting in myofibrillar degeneration and development of HCM (Hart and Cooper 1999). Both the BAG3 knockout mouse and CapZ β2 overexpression mouse illustrate the importance of CapZ in stabilization of the Z-disc and maintenance of thin filaments and reveal a role for a thin filament capping protein in HCM.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


