Fig. 36.1
Ang II-induced G protein-dependent and G protein-independent signal transduction. Ang II signaling through AT1 receptors is coupled to Gαq, activating PLC-β. This leads to generation of IP3 and DAG, resulting in vascular/cardiac contraction, protein synthesis, fibrosis, and hypertrophy. Ang II-AT1R-Gαq signals causes activation of PKC, ERK1/2, tyrosine kinases (Src, Pyk2, Tyk2), and generation of reactive oxygen species, which might activate metalloprotease ADAM-17, cleaving proHB-EGF to HB-EGF, resulting in EGFR transactivation. This initiates a MAPK signaling, leading to protein synthesis, VSMC migration, hypertrophic growth, and vascular remodeling. AT1 receptor-associated coupling to Gα12/13 activates Rho-kinase, resulting in vascular contraction and hypertrophy. The GPCR kinase-mediated receptor phosphorylation increases the receptor affinity for β-arrestin, leading to functional uncoupling of G protein signaling and also activation of cytoprotective/cardioprotective signals via p42/44 MAPK and Akt/PKB/eNOS. JAK/STAT signals can be activated by Ang II-AT1R coupling by G protein-dependent and G protein-independent mechanisms. ADAM17 A disintegrin and metalloprotease 17, Ang II angiotensin II, Akt protein kinase B, AOG angiotensinogen, AT 1 R angiotensin-II AT1 receptor, DAG diacylglycerol, EGFR epidermal growth factor receptor, eNOS endothelial nitric oxide synthase, ERK 1/2 p42/p44 mitogen-activated protein kinase or extracellular signal-regulated protein kinases 1 and 2, GPCR kinase G protein-coupled receptor kinase, GPCRs G protein-coupled receptors, HB-EGF heparin-binding endothelin growth factor, IP 3 inositol trisphosphate, JAK janus kinase, PA phosphatidic acid, PIP2 phosphatidylinositol-4,5-bisphosphate, PKC protein kinase C, PLC phospholipase C, ROS reactive oxygen species, SHP-2 Src homology phosphatase-2, SR sarcoendoplasmic reticulum, STAT signal transducer and activators of transcription, VSMC vascular smooth muscle cells
The AT1 receptor activation transduces G protein-dependent and G protein-independent signals, which in turn activate multiple downstream signaling cascades, resulting in contraction, endothelial dysfunction, cell survival and migration, hypertrophy, fibrosis, and thrombosis. AT1 receptors are coupled to Gαq/11 that stimulates the phospholipase C-protein kinase C (PLC-PKC) pathway. The AT1 receptor-mediated activation of Gβγ subunits stimulates both PLC and PLD. The latter is an important inducer of Ang II-induced oxidative stress, playing a critical role in redox-sensitive growth in human VSMCs. The PLD activation via phosphatidic acid (PA) release activates NADPH oxidase in VSMCs. Ang II-PLD-NADPH oxidase and ROS cascades function as second messengers in long-term Ang II-associated cell growth. Ang II-induced AT1 receptor interaction also activates PLA2, resulting in the production of arachidonic acid, which is metabolized by lipoxygenases to leukotrienes. The latter also activates NADPH oxidases to generate ROS [reviewed in 8, 12, 13].
Ang II-induced activation of AT1 receptors also stimulates G protein-independent signaling pathways such as β-arrestin and Src-JAK/STAT signals [reviewed in 8]. β-Arrestin would seem to be a target in developing drugs for treating cardiovascular disorders with an objective of selectively blocking the deleterious G protein-dependent pathways and simultaneously activating beneficial β-arrestin-mediated cardioprotective signaling. Currently, TRV120027, a unique drug that combines “blockade of AT1 receptor” and “β-arrestin2 stimulating activity,” is undergoing clinical trials for the treatment of acute decompensated heart failure [14]. Figure 36.1 summarizes Ang II-induced G protein-mediated signal transduction and growth factor receptor transactivation and G protein-independent signaling leading to MAPK signaling activation by the AT1 receptor.
In the subsequent section, we describe the nature and blockade of the RAAS at different upstream and downstream points that has different effects on its components. This forms the basis for describing the various classes of RAAS inhibitors for the control of blood pressure and beyond, although it does not make one class superior to another.
36.3 Drugs Targeting the RAAS Pathway
Pharmacologic agents that interrupt the functioning of the RAAS prevent the actions of Ang II either by reducing its production or by preventing its binding to target receptors. Therefore, blocking of the RAAS at different points in the axis has different effects on its components. The relative contribution of these levels in the RAAS cascade toward blood pressure regulation and prevention of cardiovascular events is not precisely known, although a considerable number of drugs interrupting the functioning of RAAS are approved and marketed worldwide. As illustrated in Fig. 36.2, these drugs fall into seven categories. The first approach, not necessarily a preferred choice, is to block the action of enzyme renin, a rate-limiting step in the synthesis of Ang I from AOG. The first such drug was aliskiren, arrived in 2007. Inhibition of this step prevents the production of angiotensin peptides by the renin-ACE and non-ACE pathways. The second approach was first employed in the late 1970s with the introduction of the ACE inhibitor, captopril. The drugs in this category have constituted one of the major advances in the treatment of mild-to-moderate hypertension, heart failure, myocardial infarction, and diabetic nephropathy. Although ACE inhibitors are well tolerated, their actions have resulted in adverse effects such as dry cough, increased bronchial reactivity, and angioedema. As the role of the RAAS in the pathophysiology of cardiovascular disease was well explored, so was the realization of the importance of finding other methods for inhibiting the actions of Ang II. This was accomplished in the third approach with the introduction of non-peptide Ang II AT1 receptor blocker (ARB), losartan, in 1995. This opened up new vistas in understanding the additional biological effects of Ang II. The AT1 receptor stimulation results in suppression of renin release. This negative feedback is interrupted where there is reduced or non-availability of angiotensin II at the receptor site, which occurs during treatment with ACE inhibitors and ARBs and leads to an increase in plasma renin activity (PRA) and plasma renin concentration (PRC). Although treatment with renin inhibitors also interrupts the negative feedback loop, resulting in elevated PRC, renin inhibitors reduce PRA and consequently decrease the generation of both Ang I and Ang II. Further downstream in the RAAS pathway, effector of Ang II, and mediator of some of its unfavorable effects, aldosterone is considered a rational therapeutic target. Thus, the fourth class, aldosterone receptor antagonists (mineralocorticoid receptor antagonists, e.g., spironolactone), was introduced for the management of hypertension and heart failure. A new and fifth strategy opened up in recent years is to inhibit the formation of aldosterone itself using aldosterone synthase inhibitors. Investigational substances blocking the enzyme aldosterone synthase are in the pipelines of pharmaceutical drug development.
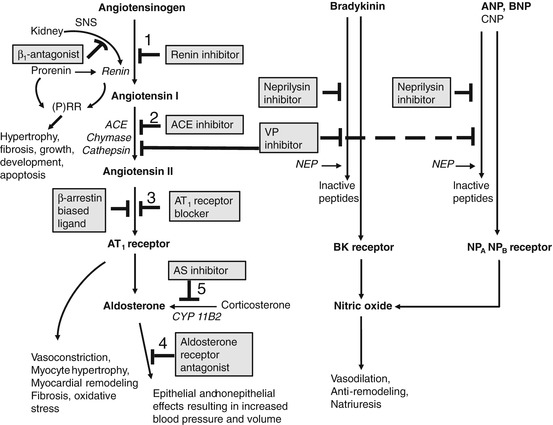
Fig. 36.2
Potential sites interrupting the functioning of RAAS are drug target for hypertension and other cardiovascular diseases. Renin converts AOG to Ang I, which further cleaved into Ang II by ACE. The cardiovascular effects of Ang II are mediated by AT1 receptors. Pharmacologic agents that interrupt the functioning of RAAS prevent the actions of Ang II by either reducing its production or preventing its binding to the AT1 receptor, thus targeting several sites in the axis involving several components. Besides the numbered potential five target sites interrupting the functioning of RAAS discussed in the text, other promising sites are also indicated in the flow diagram. β-Arrestin-biased ligand (e.g., TRV120027) combines “blockade of AT1 receptor” and “β-arrestin2 stimulating activity.” Additionally, simultaneous inhibition of AT1 receptor and NEP (e.g., LCZ696) is more promising than the inhibition of ACE and NEP (e.g., omapatrilat). Note aldosterone synthase (CYP11B2) converts 11-deoxycorticosterone to corticosterone, to 18-hydroxycorticosterone, and finally to aldosterone. Ang (1–7), a vasodilator formed from Ang II by ACE2, is not shown in the figure
Current research is focused on the development of a new class of vasopeptidase inhibitors (e.g., omapatrilat) that have a novel action of simultaneously inhibiting ACE and neutral endopeptidase (NEP, neprilysin) and thus increasing the bioavailability of natriuretic peptides. Simultaneous inhibition of both ACE and NEP has the potential to oppose the activity of the RAAS and to potentiate the vasodilatory, natriuretic, and antiproliferative effects of natriuretic peptides. This sixth approach has not met with much success because of severe adverse effects relative to ACE inhibitors, and will not be discussed further.
The seventh approach is simultaneous inhibition of the AT1 receptor and NEP. Neprilysin degrades several endogenous vasoactive peptides, including natriuretic peptides and bradykinin. Pharmacologic inhibition of neprilysin increases the levels of these vasopeptides, which could counteract the neurohormonal overactivation of Ang II and catecholamines and reduce systemic vascular resistance, sympathetic tone, vasoconstriction, sodium retention, cardiac fibrosis, ventricular hypertrophy, and maladaptive remodeling [15]. The combined inhibition of ACE and neprilysin was associated with serious angioedema. On the other hand, LCZ696, which constitutes a combination of neprilysin inhibitor sacubitril (AHU377, a prodrug that is converted to LBQ657) and an ARB (valsartan), was designed to minimize the risk of serious angioedema. A recent clinical study in chronic heart failure patients with a reduced ejection fraction compared LCZ696, an angiotensin receptor-neprilysin inhibitor (ARNI) with enalapril. In the study, LCZ696 was superior to enalapril in reducing the risks of death and hospitalization for heart failure [16]. In addition, LCZ696 was superior to enalapril in reducing the risk of death from any cause and reducing heart failure-associated symptoms and physical limitations. The LCZ696 group was noted to have higher proportions of patients with hypotension and nonserious angioedema but lower proportions with renal impairment, hyperkalemia, and cough than the enalapril group [16]. This study suggests that the combined inhibition of the AT1R and neprilysin is superior to RAAS inhibition alone in patients with chronic heart failure. The drug is still under clinical investigation.
Drugs targeting RAAS are classified as follows (Fig. 36.2):
1.
Renin inhibitor: Aliskiren
2.
ACE inhibitors: Captopril, enalapril, lisinopril, ramipril, fosinopril, benazepril, quinapril, perindopril, moexipril, trandolapril, zofenopril, and imidapril
3.
AT1 receptor blockers: Losartan, valsartan, irbesartan, eprosartan, candesartan, telmisartan, olmesartan, azilsartan, and fimasartan
4.
Mineralocorticoid or aldosterone receptor antagonists: Spironolactone and eplerenone
5.
Aldosterone synthase inhibitors: FAD 286 and LCI699 (developmental stages)
In subsequent sections, we describe drugs in each of the above class with reference to landmark clinical trials and pharmacokinetic properties followed by therapeutic uses and adverse effects. Table 36.1 summarizes the key features of approved drugs in each class.
Table 36.1
Pharmacologic, PK, therapeutic, and adverse profile of RAAS inhibitors
Drugs by site of action | Pharmacology | PK properties | Indication | Potential adverse effects | Remarks |
|---|---|---|---|---|---|
1. Renin inhibition | |||||
Aliskiren | A direct renin inhibitor. It inhibits renin and successively prevents the generation of Ang I from AOG, thereby preventing the activation of the RAAS at the proximal step in the pathway | Rapidly absorbed and reaches maximum plasma concentration within 1–3 h. High-fat meal decreases its absorption. It is eliminated in maximum through the hepatobiliary route as an unchanged drug. BA 2.5 % [23] | Treatment of hypertension | Fatigue, headache, dizziness, diarrhea, nasopharyngitis, and back pain [24]. Hypersensitivity reactions such as anaphylactic reactions and angioedema of the face and extremities | The use of aliskiren in combination with other RAASi is associated with an increased risk for hyperkalemia. Monitor serum K+ periodically. Avoid coadministration with NSAIDs. Discontinue the drug when pregnancy is detected. Discontinue the drug when pregnancy is detected |
2. ACE inhibition | |||||
Captopril | All are specific competitive inhibitor of ACE. Inhibits the conversion of angiotensin I to angiotensin II and as a result increases PRA | Rapidly absorbed and reaches maximum plasma concentration within 1 h. Food decreases its absorption. BA ~75 % | Treatment of hypertension, CHF (in combination with diuretics, digitalis), LVD after m.i., nephropathy in patients with T1DM | Hypersensitivity reactions such as anaphylactic reactions and angioedema of the face, neck, head, and extremities; cough, hyperkalemia. Black patients have higher incidence of angioedema than nonblack. Other effects are hypotension, agranulocytosis and bone marrow depression, impaired renal function, jaundice, and elevated hepatic enzymes | Dual blockade with other class of RAAS inhibitors is associated with increased risks of hypotension, syncope, hyperkalemia, changes in renal function, and acute renal failure. Do not coadminister with aliskiren in patients with diabetes and in patients with renal impairment. Avoid coadministration with NSAIDs including selective COX-2 inhibitors, which may result in deterioration of renal function/acute renal failure. The effects are usually reversible. Such coadministration may attenuate antihypertensive effect of ACE inhibitors |
Enalapril | Drug-associated angioedema might occur possibly due to accumulation of bradykinin | A prodrug bioactivated to enalaprilat. Rapidly absorbed, reaches maximum plasma concentration within 1 h. Food has no effect on absorption. BA ~60 % | Treatment of hypertension, symptomatic HF (in combination with diuretics, digitalis), asymptomatic LVD | ||
Lisinopril | Many ACE inhibitors are prodrugs. Most of them are cleared primarily by the kidney, while the impaired renal function could considerably decrease their plasma clearance. Initial dosages of all ACE inhibitors might be needed to be reduced in patients with high plasma levels of renin (HF patients and salt-depleted patients) [135] | Long acting, slow absorption, does not undergo metabolism and is excreted unchanged in the urine. Food has no effect on absorption. BA ~25 % (reduced to 16 % in CHF) | Treatment of hypertension, HF (as adjunctive therapy), acute m.i, to improve survival, diabetic nephropathy in patients with T1DM | ||
Ramipril | A prodrug hepatically cleaved to active metabolite ramiprilat. Rapidly absorbed, reaches maximum plasma concentration within 1 h. The extent of absorption (50–60 %) is not affected by the presence of food | Treatment of hypertension, cardiovascular event risk reduction, HF following m.i. | |||
Fosinopril | A prodrug with phosphinic acid binding site undergoes hepatic and gut esterification to an active drug, fosinoprilat. Slowly absorbed, reaches maximum plasma concentration, approximately 3 h. Food decreases its absorption. The BA of parent drug is ~36 % and that of metabolite is 75 % | Treatment of hypertension and in the management of HF as adjunctive therapy (added to diuretics, digitalis) | |||
Benazepril | A prodrug hepatically cleaved to active drug, benazeprilat. Rapidly absorbed, reaches maximum plasma concentration within 1 h. The extent of absorption is 37 % and. Food has no effect on absorption | Treatment of hypertension | |||
Quinapril | A prodrug quickly deesterified to its major active metabolite quinaprilat. Rapidly absorbed, reaches a maximum plasma concentration within 1 h. High-fat meal decreases the rate of absorption. BA ~60 % | Treatment of hypertension, management of HF as adjunctive therapy (added to diuretics, digitalis) | |||
Perindopril | A prodrug hydrolyzed by hepatic esterases to an active drug perindoprilat. Rapidly absorbed, reaches maximum plasma concentration within 1 h Food decreases its absorption. The BA of parent drug is 75 % and that of metabolite is 25–30 % | Treatment of hypertension, patients with stable coronary artery disease to reduce the risk of cardiovascular mortality or nonfatal m.i. | |||
Moexipril | A prodrug deesterified to its major active metabolite moexiprilat. Rapidly absorbed, reaches maximum plasma concentration in 1.5 h. Food decreases the rate of absorption. BA ~13 % | Treatment of hypertension | |||
Trandolapril | A prodrug hepatically cleaved to active metabolite trandolaprilat. Rapidly absorbed, reaches maximum plasma concentration within 1 h. Food slows absorption. BA 10 % (parent) and 70 % (metabolite) | Treatment of hypertension, HF post m.i. (in combination with diuretics, digitalis), LVD after m.i. | |||
3. AT1 receptor antagonism | |||||
Losartan | ARBs are competitive antagonists that selectively inhibit the binding of Ang II to AT1 receptors. The blockade is surmountable or insurmountable (see text for details) | It is converted in part to an active metabolite EXP 3174. Rapidly absorbed, reaches maximum plasma concentration within 1 h. Food decreases its absorption. BA ~33 % | Treatment of hypertension, to reduce the risk of stroke in patients with hypertensive and left ventricular hypertrophy (may not be applicable to Black patients) and nephropathy in T2DM with elevated serum creatinine and proteinuria | Chest pain, hypotension, hyperkalemia, hyponatremia, hypoglycemia, diarrhea, gastritis, anemia, backache, rhabdomyolysis sinusitis, and bronchitis. The incidence of cough is significantly lower than ACE inhibitor and placebo | A complete RAAS blockade is not beneficial and involves additional risks of side effects. ARBs may present an alternative to ACE inhibitors, and several trials demonstrated non-inferiority as compared to ACE inhibitors |
Valsartan | Like ACE inhibitors, ARBs inhibit negative feedback inhibition and thereby increase PRA, PRC, and Ang II. ARBs make Ang II to be available significantly at the AT2 receptor sites and activate them. However, the AT2-mediated precise beneficial actions are obscure | Reaches maximum plasma concentration in 2–4 h. Food has no effect on absorption. BA ~25 % | Treatment of hypertension and NYHA class II–IV heart failure; to reduce CV mortality in clinically stable patients with left ventricular failure or left ventricular dysfunction following m.i. (may not be applicable to Black patients); treatment of nephropathy in T2DM | Severe, chronic diarrhea with substantial weight loss has been noted in patients taking olmesartan | Do not coadminister with aliskiren in patients with diabetes and in patients with renal impairment. Avoid coadministration with NSAIDs |
Irbesartan | Rapidly absorbed, reaches maximum plasma concentration within 2 h. Food has no effect on absorption. BA 60–80 % | Treatment of hypertension and nephropathy in T2DM | ARBs also have teratogenic effect and must be avoided during pregnancy. Nursing mothers should choose to discontinue nursing or drug | ||
Eprosartan | Rapidly absorbed, reaches maximum plasma concentration within 2 h. Food delays absorption. BA ~13 % | Treatment of hypertension | An increase in cancer risk was not found with ARBs | ||
Candesartan cilexetil | Rapidly absorbed and bioactivated by ester hydrolysis in the gut. Reaches maximum plasma concentration in 3–4 h. Food does not affect the BA, which is ~15 % | Treatment of hypertension and HF (NYHA class II–IV) with left ventricular systolic dysfunction to reduce cardiovascular mortality and hospitalizations | |||
Telmisartan | Rapidly absorbed, reaches maximum plasma concentration in <1 h. Food has no effect. BA is dose dependent, 42–58 % | Treatment of hypertension and for the reduction of the risk of m.i., stroke, or death from cardiovascular causes | |||
Olmesartan medoxomil | Rapidly absorbed and bioactivated by ester hydrolysis in the gut. Reaches maximum plasma concentration in 1–2 h. Food does not affect the BA, which is ~26 % | Treatment of hypertension | |||
Azilsartan medoxomil | A prodrug rapidly absorbed and hydrolyzed in the gut. Reaches maximum plasma concentration in 1.5–3 h. Food does not affect the BA, which is ~60 % | Treatment of hypertension | |||
4. Aldosterone receptor blockade, ARAs | |||||
Spironolactone | A competitive and nonselective ARA. Causes excretion of Na+ and water while retains K+ | Rapidly and extensively metabolized. Reaches maximum plasma concentration 1–3 h. Food increases absorption. BA is ~73 %. Duration after a single dose lasts 16–24 h, and therapeutic effects may persist 2–3 days following discontinuation of therapy | Essential hypertension; primary hyperaldosteronism; edematous conditions, CHF cirrhosis of the liver accompanied by edema and/or ascites; nephrotic syndrome; hypokalemia, severe HF (NYHA class III–IV); to increase survival and to reduce the need for hospitalization | Dizziness, diarrhea, coughing, fatigue, and flu-like symptoms | Owing to “aldosterone escape phenomenon,” the escaped aldosterone (during chronic ACE inhibitors/ARBs therapy) might cause cardiovascular abnormalities, justifying the use of ARAs. This class of drug might be better than ACE inhibitors/ARBs in the treatment of low-renin hypertension and reduction in proteinuria in CKD patients. Potassium supplement, potassium-sparing diuretics, RAAS inhibitors should not be given with these drugs. Avoid coadministration with NSAIDs. Eplerenone should not be used in T2DM with microalbuminuria. Use during pregnancy only if the potential benefit justifies the potential risk to the fetus |
Eplerenone | Selectively blocks the AR with insignificant effect at other steroid receptors. Increases PRA and aldosterone by inhibiting negative regulatory feedback of aldosterone on renin secretion | Peak plasma concentration is reached in 1.5 h. Absorption is not affected by food. BA 69 %. A short half-life and does not have any active metabolites [218]. Metabolized predominantly by CYP3A4 | Hypertension, CHF following m.i. (to improve survival of stable patients with left ventricular systolic dysfunction and clinical evidence of CHF after an acute m.i.) | Both drugs can cause life-threatening hyperkalemia | |
Spironolactone causes gynecomastia, breast tenderness, erectile dysfunction, dysmenorrhea, and amenorrhea. Eplerenone has up to a 500-fold lesser affinity for androgen/progestin receptors [207, 217; thus, it causes very less progestogenic and antiandrogenic adverse effects. Eplerenone use is associated with abdominal pain and diarrhea [226] | |||||
36.3.1 Renin Inhibitors
Preprorenin is generated from renin mRNA in juxtaglomerular cells of the afferent arteriole in the kidney and is converted to prorenin. The precursor prorenin is cleaved into the active proteolytic enzyme renin. Active renin is then stored in and released from secretory granules in the kidney by exocytosis. It does so in response to a decrease in blood volume and blood pressure and increased sympathetic activity, the first step of the complex cascade of the RAAS. Prorenin is also synthesized at a high rate in a number of extrarenal tissues like the adrenal gland, collecting duct, retina, submandibular gland, placenta, ovary, and testis in several species. As the circulating levels of prorenin are much higher than those of renin, organs like the heart lacking renin expression might sequester and activate circulating prorenin [17]. Inactive prorenin is activated by irreversible proteolytic removal of the prosegment through the actions of kallikrein, convertase, trypsin, and cathepsin-B. The removal of the prosegment peptide from prorenin produces enzymatically active renin in the kidney. Also the inactive prorenin is activated by reversible nonproteolytic binding with the (pro)renin receptor (PRR) where the prosegment moves out of the catalytic site without getting detached from prorenin [reviewed in 18].
Both prorenin and renin bind to the PRR [19]. The circulating renin and prorenin bound PRR not only triggers the local generation of Ang II but also activates Ang II-independent signaling cascades. The PRR activation stimulates the phosphorylation of MAPKs, resulting in expression of profibrotic genes, generation of TGF-β1, plasminogen activator inhibitor-1, fibronectin, and collagen-1 synthesis, leading to end-organ damage [reviewed in 18]. Prorenin is no longer an inactive precursor of renin as its binding to the PRR dually activates tissue Ang II-dependent and -independent signal transduction leading to hypertension, cardiac fibrosis, glomerulosclerosis, and proteinuria. Blocking the PRR, the site strategically located at the proximal part of the RAAS cascade, should therefore lessen both the catalytic effect (Ang II generation) and noncatalytic signal transduction of renin and prorenin. At present, the logical target at a higher level in the cascade for RAAS inhibition is to block the action of the enzyme renin, a rate-limiting step in the synthesis of Ang I from AOG. This prevents the production of angiotensin peptides by renin-ACE and non-ACE pathways.
Research on renin inhibition started with antibodies and peptides even before the discovery of captopril. The first generation of renin inhibitors, peptides (e.g., peptastatin) or peptidomimetics (e.g., enalkiren, remikiren), demonstrated renin inhibitory activity and reduced blood pressure. However, they exhibited a short duration of action and low potency and had poor oral bioavailability (<2 %). The knowledge of the tridimensional structure of recombinant human renin opened the doors for structure-based design of renin inhibitors [20]. Using computational molecular modeling and crystallographic techniques, scientists synthesized a number of potent and selective non-peptidic low molecular weight renin inhibitors [21]. The lead candidate for further clinical development was aliskiren, which is the only direct renin inhibitor approved for the treatment of hypertension. Although aliskiren is well tolerated and demonstrated non-inferiority to ACE inhibitors and ARBs for blood pressure reduction, it has a relatively low bioavailability (~2.5 %) and requires high doses to achieve a similar blood pressure reduction to that of ACE inhibitors and ARBS. This prompted the development of newer renin inhibitors with improved therapeutic profiles. Currently, a number of them are under different phases of clinical trials, the most prominent among them is VTP-27999 [22].
36.3.1.1 Aliskiren
This first-in-class selective renin inhibitor was approved in March 2007 by the US Food and Drug Administration (FDA) for the management of primary hypertension. Following oral administration, aliskiren is rapidly absorbed and reaches the maximum plasma concentration within 1–3 h, while its absolute bioavailability is a meager 2.5 %. A high-fat meal substantially decreases the absorption of aliskiren. Aliskiren is eliminated through the hepatobiliary route as unchanged drug and, to a lesser extent, through oxidative metabolism. About 0.6 % of the dose is recovered in the urine [23].
Aliskiren is indicated for the management of patients with mild-to-moderate hypertension who are intolerant to first-line antihypertensive therapies [24]. A recent meta-analysis from clinical studies suggested that the blood pressure control in hypertensive patients was better with aliskiren when combined with either amlodipine or hydrochlorothiazide than alone, with aliskiren and amlodipine combination being more effective than aliskiren and hydrochlorothiazide combination [25]. The Aliskiren in Left Ventricular Hypertrophy (ALLAY) trial compared the effects of aliskiren and losartan, alone and in combination, on left ventricular mass index in hypertensive subjects with left ventricular hypertrophy [26]. For a similar blood pressure reduction, aliskiren was as effective as losartan in promoting left ventricular mass regression. Additionally, the reduction in left ventricular mass index in the combination group was not significantly different from that of losartan alone [26]. The Aliskiren Observation of Heart Failure Treatment (ALOFT) study in patients with New York Heart Association (NYHA) class II–IV heart failure suggested that addition of aliskiren to an ACE inhibitor (or ARB) and beta-blocker appeared to be well tolerated and had favorable neurohumoral effects [27]. The Aliskiren in Evaluation of Proteinuria I Diabetes (AVOID) trial in patients with diabetes, hypertension, and diabetic nephropathy suggested that aliskiren might have renoprotective effects independent of its blood pressure-lowering effect [28]. Likewise, the combination of aliskiren and irbesartan was more antiproteinuric than monotherapy in type 2 diabetic patients with albuminuria [28]. The ASPIRE (Aliskiren Study in Post-MI Patients to Reduce Remodeling) trial that included patients ∼2 and 8 weeks following acute myocardial infarction with the left ventricular ejection fraction (LVEF) ≤45 % compared the administration of increasing doses of aliskiren with a placebo in patients receiving standard treatment with an ACE inhibitor or an ARB. Aliskiren was no more effective than was placebo at reducing the cardiovascular endpoints. Adding aliskiren to the standard therapy, including an inhibitor of the RAAS, in high-risk post-myocardial infarction patients did not result in further attenuation of left ventricular remodeling. On the other hand, aliskiren in this group of patients statistically significantly increased the risk of renal failure, hypotension, and hyperkalemia [29]. The marked increase in risk overshadowed a nonsignificant trend toward a benefit associated with cardiovascular endpoints. Additionally, the Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE) study in patients of type 2 diabetes mellitus and chronic kidney disease, cardiovascular disease, or both showed that the addition of aliskiren to an ACE inhibitor or an ARB is harmful as demonstrated by higher risk of renal impairment, stroke, hypotension, and hyperkalemia [30]. The trial was terminated early. The combined RAAS blockade strategy therefore may not be beneficial in hypertensive patients with metabolic disorders.
36.3.1.2 Therapeutic Use
Aliskiren is indicated for the management of primary hypertension to lower blood pressure, which could reduce the risk of fatal and nonfatal cardiovascular events like stroke and myocardial infarction.
36.3.2 ACE Inhibitors
ACE (dipeptidyl carboxypeptidase, also termed kininase II) is a nonspecific enzyme that cleaves dipeptide units from substrates with diverse amino acid sequences, especially Ang I and bradykinin. ACE inhibitors are structurally heterogeneous. They differ in the specific groups that bind to ACE, and this influences their pharmacokinetics. A study on the comparative binding affinity of some ACE inhibitors for the two binding sites of human endothelial ACE revealed that ACE inhibitors generally had higher affinity for bradykinin than for Ang I binding sites, suggesting that these agents are primarily, inhibitors of bradykinin degradation, and secondarily, inhibitors of Ang II production [31]. Among those tested, perindoprilat had the highest selectivity for bradykinin versus Ang I binding sites, while enalaprilat had the lowest, indicating that there are differences in the action of ACE inhibitors toward their substrates [31]. This could lead to differences in their efficacy in the treatment of cardiovascular disease.
Different drugs of this class exhibit different hydrophilic and lipophilic properties resulting in different ability to penetrate into tissues. Currently more than 12 ACE inhibitors are in use worldwide. They fall into three classes based on their chemical structures:
1.
Sulfhydryl-containing agents: captopril and zofenopril
2.
Carboxylate-containing agents: enalapril, ramipril, quinapril, perindopril, lisinopril, benazepril, imidapril, trandolapril, and moexipril
3.
Phosphonate-containing agent: fosinopril
Zofenopril and imidapril are not available in the USA.
Although all ACE inhibitors are approved for the treatment of hypertension, all are not equally effective for all indications. Quantitative differences exist among ACE inhibition. Some of ACE inhibitors are lipophilic (e.g., fosinopril >>>quinapril = enalapril > captopril = ramipril = perindopril) that might influence enzyme-binding capabilities with tissue ACE [32]. This can theoretically provide greater penetration into the atherosclerotic plaque. However, this unique lipophilicity has not been shown to predict clinical outcomes [33]. A case-control study has shown that the risk of nonfatal myocardial infarction (MI) did not correlate with the degree of ACE inhibitor lipophilicity (tissue affinity) [33, 34]. However, in a randomized study involving patients who had an MI [35], high-tissue penetrating quinapril was shown to be significantly more effective than was comparatively low-tissue penetrating enalapril in reducing the concentration of C-reactive protein. One retrospective study demonstrated differential survival benefits in the first year after acute MI in patients 65 years of age or older to the specific ACE inhibitor prescribed, while ramipril was associated with lower mortality than were most other ACE inhibitors like enalapril, fosinopril, captopril, quinapril, and lisinopril [32]. This study suggested that ACE inhibitors do not benefit acute MI patients similarly. The differential efficacy among specific ACE inhibitors appears to extend to coronary artery disease (CAD) patients without a history of MI, as reported for ramipril in the HOPE study and for perindopril in the EUROPA study. On the other hand, trandolapril in the PEACE (Prevention of Events with Angiotensin-Converting Enzyme Inhibition) trial with stable CAD demonstrated no reduction in the primary composite endpoint of CV death and MI. The findings from these trials suggest that a “class effect” may not exist among ACE inhibitors for the treatment of stable CAD [36].
36.3.2.1 Mechanism of Action
Structurally, ACE is a zinc metalloprotease with two active catalytic sites: NH2 and COOH sites. ACE inhibitors bind to the active site of ACE and interfere with the ability of the enzyme to bind and cleave its substrates. As a result, the cleavage of Ang I to Ang II is inhibited. This results in reduced availability of Ang II at the receptor sites. Bradykinin, another substrate for ACE and naturally occurring vasodilator, is also prevented from its degradation by ACE inhibitors. Consequently, bradykinin binds to endothelial bradykinin B2 receptors resulting in nitric oxide-mediated vasodilation and associated effects. ACE inhibitors increase PRA and PRC by interfering with the negative feedback loop. This process results in increased formation of Ang I, which may be converted by neutral endopeptidase (NEP) to Ang (1–7), a vasodilator. However, it is not clear to what extent Ang (1–7) can contribute to alleviating clinical hypertension during ACE inhibitor therapy. ACE inhibitors decrease systemic vascular resistance without increasing heart rate [37]. Most ACE inhibitors are cleared principally by the kidney, so impaired renal function could significantly lessen the plasma clearance of these agents. The dose of ACE inhibitors should therefore be reduced in patients with impaired renal function.
36.3.2.2 Heterogeneity of the ACE Gene May Influence the Effectiveness of ACE Inhibitors
The variations in response to ACE inhibitors might be associated with ACE insertion/deletion (I/D) polymorphism [38]. The ACE gene polymorphism is based on the presence or absence of a 287-bp element on intron 16 on chromosome 17 [38]. The level of circulating ACE enzymes depends on the I/D polymorphism [39]. These differences in plasma ACE activity associated with the ACE genotype might modify the efficacy of ACE inhibitors. Patients with a deletion genotype at the intron 16 of the ACE gene exhibited higher activity of plasmatic ACE compared to patients with the insertion genotype [40]. Hass et al. [40], in patients with biopsy-proven proteinuric glomerular diseases and the DD (n = 10) and ID/II (n = 26) genotype, studied the hemodynamic and antiproteinuric effect of a 6-month therapy using enalapril. Neither blood pressure nor proteinuria changed significantly in patients with the DD genotype in spite of slightly higher dose of enalapril. In contrast, both were significantly reduced in the II/ID group after 10 weeks and 6 months of enalapril therapy. Creatinine clearance was noted to be decreased steadily in DD patients. On the other hand, in II/ID patients, creatinine clearance was noted to be reduced significantly after 10 weeks of enalapril therapy, and then it increased again, while the value at 6 months was again similar to that of DD patients [40]. This study concluded that ACE genotype could influence the antihypertensive and antiproteinuric effect of enalapril in patients with proteinuric glomerular disease [40]. Of note, Kohno et al. [41] suggested that hypertensive patients with the DD genotype when treated with ACE inhibitors were less likely to have regression of left ventricular hypertrophy than were patients with other ACE genotypes [41]. In this study, 18 patients of II genotype for the ACE gene, 19 patients of ID genotype, and 17 patients of DD genotype were tested. Baseline serum ACE activity was significantly greater in the DD group than in the II or ID genotype groups. Despite similar blood pressure reductions, after 2 years, mean regression in posterior wall thickness was significantly less in the DD group than in the ID and II genotype groups [41]. In a randomized placebo-controlled trial of perindopril in normotensive, normoalbuminuric patients with type 1 diabetes mellitus, the nephroprotective effect of ACE inhibition was not associated with the ACE genotype (II, ID, DD) [42]. In a study of 479 patients with systolic dysfunction (LVEF 0.25 ± 0.08) who were genotyped for the ACE-D/I polymorphism [43] and followed to the endpoint of death or cardiac transplantation, 227 patients received ACE inhibitors at “low doses” (≤50 % of target dose), 201 patients received “high (standard) dose,” and 51 patients received ARBs. The ACE-D allele was noted to be associated with an increased risk of events while this effect was primarily in the low-dose group (1-year percent event-free survival: II/ID/DD = 86/77/71, 2-year = 79/66/59, p = 0.032). In the standard-dose group, the impact was noted to be markedly diminished (1-year: II/ID/DD = 91/81/80, 2-year: 77/70/71, p = 0.64). The impact of beta-blockers and high-dose ACE inhibitors was noted to be greatest in patients of ACE DD genotype and was less apparent with II and ID genotypes [43], suggesting that determination of ACE genotype might help target therapy for heart failure. Evidence also highlights that ACE gene DD polymorphism is associated with poorer survival and an increase in left ventricular mass in idiopathic heart failure patients, suggesting a possible pathophysiologic pathway between ACE gene polymorphism, ACE activity, myocardial hypertrophy, and survival [44]. Taken together, ACE gene polymorphism might play a key role in a marked interindividual difference in the efficacy of ACE inhibitors. However, further studies are needed for a confirmation (Chap. 35).
36.3.2.3 Captopril
It was the first-in-class short-acting ACE inhibitor approved in 1981. After oral administration, captopril is absorbed rapidly with a bioavailability of around 75 %. It is eliminated in urine, 40–50 % as captopril and the rest as captopril disulfide dimers and captopril-cysteine disulfide. Ingestion of food decreases its absorption and bioavailability [45]. The Survival and Ventricular Enlargement (SAVE) trial examined the effect of captopril in patients within 3–16 days after myocardial infarction, with ejection fraction ≤40 % and without overt heart failure or symptoms of myocardial ischemia [46]. In patients with asymptomatic LV dysfunction after MI, long-term treatment with captopril improved survival and reduced morbidity and mortality due to cardiovascular events, and these benefits were seen in patients who received thrombolytic therapy, aspirin, or β-blockers as well as those who did not [46]. The Captopril Prevention Project (CAPP) evaluated the effects of an ACE inhibitor-based therapeutic regimen on cardiovascular events in hypertensive diabetic patients. In the study, captopril was superior to a diuretic/beta-blocker in preventing cardiovascular events in hypertensive diabetic patients, particularly in those with metabolic decompensation [47]. In addition, a captopril-based antihypertensive treatment regimen was reported to be associated with a lower risk of diabetes mellitus development when compared with conventional therapy based on diuretics and/or beta-blockers [48]. Captopril is also shown to reduce progression of diabetic nephropathy. The European Microalbuminuria Captopril Study in non-hypertensive patients with insulin-dependent diabetes mellitus and persistent microalbuminuria revealed that captopril therapy significantly impeded the progression to clinical proteinuria and prevented the increase in albumin excretion rate [49]. Likewise, the North American Microalbuminuria Study in normotensive subjects with insulin-dependent diabetes mellitus reported that 24 months of therapy with captopril was well tolerated, and, compared to placebo, it reduced significantly the progression of microalbuminuria to clinical proteinuria and also the albumin excretion [50]. The Captopril in Heart Insufficient Patients Study (CHIPS) in patients with mild-to-moderate heart failure indicated that a high dose of captopril as compared to a low dose improved the long-term clinical outcome without significantly increased toxicity [51]. In the study of Kazerani et al. [52], sublingual captopril was reported effective, easily applicable, and safe treatment for the management of hypertensive urgency for 120 min for those patients who did not receive a multidrug antihypertensive regimen. A recent study showed a similar effect of sublingual and oral captopril in hypertensive crisis [53]. However, the taste of the sublingual drugs might be unpleasant.
36.3.2.4 Enalapril
Enalapril is a relatively inactive prodrug with good oral absorption (60–70 %). It is hydrolyzed by esterases in the liver to form enalaprilat, which is a potent ACE inhibitor [54]. Most of the drug undergoes renal elimination as either intact enalapril or enalaprilat. Since enalaprilat as such is not absorbed orally, it is available for intravenous administration. Chronic hypertension considerably increases cardiovascular disease risk in patients with diabetes mellitus. The Appropriate Blood Pressure Control in Diabetes (ABCD) trial in type 2 diabetic hypertensive patients over a 5-year follow-up period demonstrated an advantage of enalapril over nisoldipine (a long-acting calcium channel antagonist) in reducing the incidence of cardiovascular events [55]. Nisoldipine was noted to be associated with a higher incidence of fatal and nonfatal myocardial infarctions than was enalapril in patients with type 2 diabetes mellitus and hypertension [56]. In the studies of Jong et al. [57], treatment with enalapril for 3–4 years in patients with left ventricular systolic dysfunction led to a sustained improvement in survival beyond the original trial period. In patients with heart failure, both enalapril and fosinopril have similar short-term effects on event-free survival, ejection fraction, functional capacity, and quality of life [58]. In patients on hydrochlorothiazide with uncontrolled blood pressure, enalapril was more effective than amiloride in lowering blood pressure [59]. In patients with mild-to-moderate hypertension, a fixed-dose combination of enalapril and nitrendipine was well tolerated and effectively lowered blood pressure, with lower incidence of edema than with calcium channel blocker monotherapy [60, 61]. The Studies Of Left Ventricular Dysfunction (SOLVD) trial in patients with congestive heart failure reported that diabetes mellitus was associated with an increased risk of renal impairment, but this risk was reduced in the enalapril group compared with the placebo group [62]. In a recent clinical study in stable, older patients with compensated heart failure and preserved ejection fraction and controlled blood pressure, enalapril treatment for 12 months failed to improve exercise capacity or aortic distensibility, suggesting that ACE inhibition might not substantially improve key long-term clinical outcomes in this group of patients [63].
36.3.2.5 Lisinopril
On oral administration, lisinopril is absorbed slowly and incompletely and, thus, is a long-acting ACE inhibitor. Food has no significant effect on the absorption of lisinopril. The extent of absorption is approximately 25 %. However, its bioavailability is reduced to about 16 % in patients with stable NYHA class II–IV CHF [see 64, US FDA labeling for lisinopril]. It does not undergo metabolism, and it is excreted intact in the urine [65]. Impaired renal function decreases the elimination of lisinopril. In patients with mild-to-moderate essential hypertension, lisinopril is well tolerated and effectively reduces the blood pressure [66]. In type 1 and type 2 diabetic hypertensive patients and early or overt nephropathy, lisinopril lowers blood pressure and preserves renal function without adversely affecting glycemic control or lipid profiles [67, 68]. The EUrodiab Controlled trial of Lisinopril in Insulin-Dependent Diabetes (EUCLID) trial supported an additional place for lisinopril in managing normotensive patients with type 1 diabetes mellitus and microalbuminuria [67]. Furthermore, it was noted that hypoglycemia occurred at a similar frequency in both lisinopril and placebo groups, while patients on lisinopril had significantly lower HbA1c at baseline than those on placebo [67, 69]. The Italian Microalbuminuria Study in normotensive insulin-dependent diabetes mellitus patients with microalbuminuria showed that lisinopril was effective in delaying the occurrence of macroalbuminuria [70]. Lisinopril appeared powerful in slowing the course of nephropathy [70]. The Blood Pressure Reduction and Tolerability of Valsartan in Comparison with Lisinopril (PREVAIL) study in patients with mild-to-severe hypertension reported that both lisinopril and valsartan were highly effective in controlling blood pressure, while valsartan was noted to be associated with a substantially reduced risk for cough [71]. The fixed combination of lisinopril and hydrochlorothiazide in patients with essential hypertension was efficacious in treating essential hypertension and showed a regression of left ventricular hypertrophy [72, 73]. A recent study (DETECT trial) investigated the effects of carvedilol, lisinopril, and their combination for 9 months on vascular and cardiac health in patients with borderline blood pressure. In this study, all treatment groups produced a sustained and well-tolerated functional improvement but not a structural improvement [74].
36.3.2.6 Ramipril
Ramipril is transformed by hepatic esterases into ramiprilat. After oral administration, ramipril is rapidly absorbed, which is reduced in the presence of food. The glucuronides of ramipril and ramiprilat undergo renal excretion. The Acute Infarction Ramipril Efficacy (AIRE) trial in patients with clinical evidence of either transient or ongoing heart failure reported that oral administration of ramipril, initiated between the second and ninth day after myocardial infarction, substantially reduced premature death from all causes, and this benefit was apparent as early as 30 days and was consistent across a range of subgroups [75]. The Ramipril Efficacy In Nephropathy (REIN) study in patients with chronic nephropathies and high proteinuria reported that ramipril safely reduced the rate of decline of the glomerular filtration rate and halved the risk of end-stage renal failure [76]. The REIN follow-up study in patients with chronic nephropathy and high risk of rapid progression to end-stage renal failure showed that ramipril reversed the tendency of glomerular filtration rate to decline with time [76]. In addition, patients previously treated with antihypertensive drugs other than ACE inhibitors benefited from shifting to ramipril [76]. The effects of ramipril on coronary events in high-risk persons were evaluated in the Heart Outcomes Prevention Evaluation (HOPE) trial. The study reported that in high-risk cohort, ramipril reduced the risk of myocardial infarction, worsening and new angina, and the occurrence of coronary revascularizations [77]. In addition, ramipril reduced the risk of fatal and nonfatal serious arrhythmic events in high-risk patients without clinical heart failure or overt left ventricular systolic dysfunction [78]. A recent clinical study showed that ramipril improved walking distance in patients with claudication possibly through reduction of arterial stiffness [79]. Likewise, among patients with intermittent claudication, 24-week treatment with ramipril significantly increased pain-free and maximum treadmill walking times relative to those taking placebo [80].
36.3.2.7 Fosinopril
Fosinopril is a phosphate-containing prodrug transformed by hepatic esterases into fosinoprilat. This metabolite is more potent than captopril but less potent than enalaprilat. After oral administration, fosinopril is absorbed slowly and incompletely and averages 36 % of an oral dose. Both fosinoprilat and the glucuronide conjugate of fosinoprilat are excreted in urine and bile [81]. The Fosinopril Versus Amlodipine Cardiovascular Events Randomized Trial (FACET), which compared the effects of fosinopril and amlodipine on serum lipids and glucose control in patients with type 2 diabetic patients with hypertension, showed both treatments were equally effective in lowering blood pressure. There were no significant differences in lipid profile and glucose control between the two groups. However, patients receiving fosinopril had a significantly lower risk of combined outcome of acute myocardial infarction, stroke, or hospitalized angina than those receiving amlodipine alone [82]. Fogari et al. [83] compared the long-term effect of the combination of amlodipine and fosinopril versus monotherapy on urinary albumin excretion in hypertensive diabetic patients. In this study, combination therapy was more effective in reducing blood pressure than either drug alone at any time of the study without affecting glucose homeostasis. Although all three treatments, amlodipine, fosinopril, or their combination, caused a significant decrease in urinary albumin excretion during the 48-month study period, the combination had a greater effect on reducing albuminuria than either drug did alone [83].
36.3.2.8 Benazepril
Benazepril is a prodrug and is converted by hepatic esterases into benazeprilat, which is a potent inhibitor of ACE. Food slightly delays the absorption of benazepril, but may not affect its ultimate bioavailability. Severe hepatic impairment could slow the conversion of benazepril to benazeprilat, but may not affect the overall bioavailability of benazeprilat. Benazepril is mostly metabolized to benazeprilat and benazepril-glucuronide conjugates [84, 85]. The Avoiding Cardiovascular Events in Combination Therapy in Patients Living with Systolic Hypertension (ACCOMPLISH) trial in patients with hypertension who were at high risk for cardiovascular events reported that the combination of benazepril and amlodipine was superior to the combination of benazepril and hydrochlorothiazide in reducing cardiovascular events [86]. It was further reported that initial antihypertensive treatment with the combination of benazepril and amlodipine should be considered in preference to the combination of benazepril and hydrochlorothiazide since it slowed the progression of nephropathy to a greater extent [87]. Likewise, a recent clinical trial reports the non-inferiority of the combination of amlodipine and benazepril, as compared to the combination of valsartan and hydrochlorothiazide, in lowering blood pressure [88]. Moreover, this combination exerted beneficial effects on renal function, glucose control, and HDL-C and triglyceride levels when compared with the combination of valsartan and hydrochlorothiazide [88].
36.3.2.9 Quinapril
Quinapril is a prodrug transformed by hepatic esterases into quinaprilat, a long-acting ACE inhibitor [89]. After oral administration, quinapril is rapidly absorbed. The metabolite quinaprilat is excreted in urine and feces. Diminished liver function could reduce the transformation of quinapril to quinaprilat. In patients with hypertension, the efficacy of quinapril was similar to that of other ACE inhibitors and showed a lower incidence of adverse events or withdrawals for adverse events relative to those associated with captopril or enalapril [90]. The TREND (Trial on Reversing ENdothelial Dysfunction) study in normotensive patients with coronary artery disease showed that ACE inhibition with quinapril improved endothelial dysfunction [91]. Interestingly, quinapril substantially increased the production of total nitric oxide relative to enalapril following acute myocardial infarction [92].
Comparative trial of quinapril versus captopril in patients with mild-to-moderate congestive heart failure suggested that treatment with 20 mg quinapril once a day was as effective as 100 mg captopril twice a day [93]. The results of this study are generally consistent with those of Acanfora et al. [94] who showed quinapril was as effective as captopril in reducing signs and symptoms of congestive heart failure patients (New York Heart Association [NYHA] class II to III) and improving left ventricular function and exercise capacity. In the study of Zi et al. [95], quinapril did not show beneficial effects on exercise tolerance and quality of life in elderly heart failure patients with preserved systolic function. The Sadko-CHF study in patients with mild-to-moderate congestive heart failure showed no significant benefit with triple combination (quinapril, valsartan, and bisoprolol) over the combination of quinapril and bisoprolol or bisoprolol and valsartan in the functional status, quality of life, and parameters of left ventricular remodeling [96]. Furthermore, the combination of quinapril and bisoprolol was more effective on 24-h heart rate variability parameters, sympathoadrenal activity, and renal function than the combination of bisoprolol and valsartan or triple combination [96]. In this study, the triple combination might have a negative effect on neurohormonal profile such as excessive activation of Ang II and epinephrine. Thus, the investigators did not recommend the triple combination therapy for stable mild-to-moderate congestive heart failure patients [96]. A recent study in patients with arterial hypertension and functional class I–II chronic heart failure with preserved left ventricular ejection fraction demonstrated the advantages of quinapril monotherapy over metoprolol therapy [97]. Quinapril treatment has also been shown to improve insulin resistance and low-grade inflammatory state in hypertensive patients [98].
36.3.2.10 Perindopril
It is a non-sulfhydryl ester prodrug transformed by hepatic esterases to its active diacid metabolite perindoprilat. It is a long-acting ACE inhibitor. The major part of the active metabolite is cleared by the kidneys, while the other major metabolite of perindopril is an inactive glucuronide. Aging is associated with increased serum perindoprilat concentration, which is possibly caused by a combination of enhanced conversion to the active metabolite and diminished renal clearance [99]. Perindopril was reported to normalize blood pressure in a large majority of hypertensive diabetic patients. In patients at risk of developing diabetic nephropathy, perindopril induced a marked and sustained reduction of microalbuminuria [100]. In the study of Hui et al. [101], perindopril induced a significant regression of left ventricular hypertrophy associated with improvement in left ventricular diastolic performance. Furthermore, perindopril was more effective than was metoprolol in reversing left ventricular hypertrophy [101]. In the EUropean trial on Reduction Of cardiac events with Perindopril in patients with stable coronary Artery disease (EUROPA) study in patients with stable coronary heart disease and no apparent heart failure, perindopril significantly improved the cardiovascular outcome. Additionally, the investigators recommend considering treatment with perindopril, on top of other preventive medications, in all patients with coronary heart disease [102]. Similar results were reported in the PERSUADE study of Daly et al. [103], in which perindopril tended to reduce major cardiovascular events in diabetic patients with coronary artery disease. According to Ferrari et al. [104], the direct vascular protective property of perindopril might be a reason for its beneficial action in preventing cardiac events in stable coronary artery disease patients. The ASCOT-BPLA trial showed that the free combination of amlodipine and perindopril effectively controlled blood pressure and was superior to the combination of beta-blocker/diuretic in reducing total mortality and cardiovascular outcomes [105]. The SafeTy & efficacy analysis of coveRsyl amlodipine in uncOntrolled and Newly diaGnosed hypertension (STRONG) study concluded that the fixed combination of perindopril and amlodipine was an effective and well-tolerated antihypertensive treatment [105]. Results from a recent clinical study reveal that the addition of a fixed-dose combination of perindopril and amlodipine to blood pressure regimen is efficient in terms of blood pressure control for 62.3 % of those patients with not-at-goal hypertension [106]. The Action in Diabetes and Vascular disease: PreterAx and DiamicroN-MR Controlled Evaluation (ADVANCE) trial showed that routine administration of a fixed-dose combination of perindopril and indapamide to patients with type 2 diabetes mellitus was well tolerated and reduced the risks of major vascular events [107]. The trial also showed that the systematic use of a fixed-dose combination of perindopril and indapamide afforded substantial protection against cardiovascular mortality and myocardial infarction and also renoprotection by reducing the development of micro- and macroalbuminuria [108].
36.3.2.11 Moexipril
Moexipril is a prodrug hydrolyzed after oral administration to its active metabolite moexiprilat [109]. Its bioavailability is markedly reduced in the presence of food and thus should be taken in a fasting state. Patients with moderate-to-severe hypertension inadequately controlled with hydrochlorothiazide benefitted from the addition of moexipril [110]. A statistically greater reduction in left ventricle mass was observed when moexipril was added to a diuretic than to a calcium channel blocker or a beta-blocker [111]. In hypertensive patients, moexipril monotherapy reversed left ventricular hypertrophy [112]. The MOexipril and REgression of left ventricle hypertrophy in combination therapy (MORE) trial reported a greater effect on blood pressure reduction with a combination of moexipril and diuretic than the combination of an ACE inhibitor and a beta-blocker or an ACE inhibitor and a calcium channel blocker [111].
36.3.2.12 Trandolapril
After oral administration, trandolapril is bioavailable as trandolapril (less extent) and trandolaprilat (more extent). The metabolite is about eight times more potent than is the parent compound in inhibiting ACE activity. Trandolaprilat and its inactive metabolites like glucuronides of trandolapril and deesterified products are recovered in the urine and feces. The plasma clearance of trandolaprilat is diminished by renal and hepatic insufficiency [see 113 for FDA labeling]. In the Danish TRACE study (Trandolapril Cardiac Evaluation) in patients with left ventricular dysfunction soon after myocardial infarction, trandolapril markedly reduced the risk of overall mortality, mortality from cardiovascular causes, sudden death, and the development of severe heart failure [114]. A titration-based, escalating-dose regimen of trandolapril was suggested to be effective and well tolerated in the management of subjects who were antihypertensive treatment naive or whose disease was uncontrolled on a diuretic or calcium channel blocker [115]. The fixed-dose combination of trandolapril and verapamil SR in patients with hypertension including those with type 2 diabetes mellitus was more effective than monotherapy [116]. The International Verapamil-Trandolapril Study (INVEST) trial in hypertensive coronary artery disease patients reported that the verapamil-trandolapril-based strategy was as clinically effective as was the atenolol-hydrochlorothiazide-based strategy [117].
36.3.2.13 Therapeutic Uses of ACE Inhibitors
(i)
Treatment with ACE inhibitors is beneficial for the management of patients with mild-to-moderate hypertension. Most of hypertensive patients require at least two antihypertensive drugs to achieve optimal blood pressure control, while the use of combination therapy as first-line treatment is highly recommended. Patients with mild-to-moderate hypertension can better be managed with a combination of an ACE inhibitor and a Ca2+ channel blocker or a β-adrenergic receptor blocker or a diuretic than alone.
(ii)
ACE inhibitors may be more suitable for the management of blood pressure in hypertensive patients with diabetes mellitus because of improvement of endothelial function and reduction of cardiovascular events.
(iii)
ACE inhibitors are a valuable class of drugs for the management of patients with systolic dysfunction because they prevent/delay the progression of heart failure (see Box 36.1) and decrease the incidence of myocardial infarction and hospitalization. Therapy with ACE inhibitors might reduce overall mortality given that the treatment begins during the peri-infarction period. In patients with high risk of cardiovascular events, treatment with ACE inhibitors results in a decrease in the rate of myocardial infarction (Chaps. 8 and 20).
(iv)
Treatment with ACE inhibitors prevents/delays the progression of nephropathy in type 1 diabetic as well as nondiabetic patients by reducing glomerular capillary pressure and glomerular injury.
(v)
Both American and European guidelines for the management of stable angina, acute coronary syndromes, or ST-elevation myocardial infarction provide a broad recommendation for the use of ACE inhibitors (or ARBs, if patients are intolerant to ACE inhibitors) in patients with coronary artery disease [55, 118–125] (Chap. 20).
A substantial number of hypertensive patients are not adequately controlled with ACE inhibitors (e.g., Afro-Americans show poor response because of low levels of renin). ACE inhibitors might be less effective in heart failure patients with high level of epinephrine [126]. For adequate blood pressure control in these patients, a combination with diuretics, beta-blockers, and/or calcium channel blocker is recommended.
36.3.2.14 Adverse Effects of ACE Inhibitors
ACE inhibitors are not associated with serious untoward reactions. Their use can cause reversible hypotension. Incidence of hyperkalemia (in hypertensive patients with renal insufficiency or under treatment with K+-sparing diuretics) is reported in high percentage of patients taking ACE inhibitors. Head and neck angioedema (up to 0.7 % of patients) characterized by rapid swelling in the skin of the face, around the mouth, and the mucosa of the mouth and/or throat, as well as the tongue occurs usually within the first week of therapy or within the first few hours after the initial dose. Angioedema and dry cough (5–35 % patients), the class effects of ACE inhibitors, occur possibly because of ACE inhibition-associated accumulation of bradykinin. However, these might disappear on cessation of therapy with ACE inhibitors. They are more common among African-Americans/Afro-Caribbeans (life threatening in 20 % cases). Acute renal failure while on ACE inhibitor therapy is not uncommon. ACE inhibitor-induced hepatotoxicity is rare with an incidence less than 0.1 %. Most hepatotoxicity is mild and transient. Enalapril, fosinopril, lisinopril, and ramipril have been linked to overt hepatotoxicity. As a class effect, ACE inhibitors can cause a fetal risk if taken during pregnancy. Fetal and neonatal morbidity and death have occurred from the use of these drugs during the term of pregnancy [37, 127–135]. All classes of drugs inhibiting RAAS carry a boxed warning on the label to discontinue when pregnancy is detected (see Box 36.2). Arrhythmias including both ventricular and atrial tachycardia, atrial fibrillation, premature ventricular contractions, and bradycardia may occur with drugs like lisinopril [136]. Other adverse effects of ACE inhibitors are skin rash usually consisting of a pruritic maculopapular eruption (less frequently); dysgeusia as characterized by an alteration in or loss of taste, which might often be associated with the use of captopril (the sulfhydryl drug); neutropenia (rarely but predominantly occurs in hypertensive patients with collagen-vascular or renal parenchymal disease) [135]; and sudden and potentially life-threatening anaphylactoid reaction (e.g., with lisinopril) in some patients undergoing dialysis [137].
Box 36.1: ACE Inhibitors in Heart Failure
In patients with heart failure, clinical trials demonstrated the survival benefits of ACE inhibitors (Chap. 8). However, ACE inhibitors are sometimes underutilized in these patients because of renal insufficiency or the rise in serum creatinine level. It is suggested that ACE inhibitor therapy might not be discontinued unless serum creatinine level rises above 30 % over baseline during the first 2 months after initiation of therapy or development of hyperkalemia (serum potassium level ≥5.6 mmol/L) [138].
Box 36.2: Prescribing Information: Boxed Warning from the US FDA
WARNING: FETAL TOXICITY
See full prescribing information for complete boxed warning.
When pregnancy is detected, discontinue [DRUG] as soon as possible. Drugs that act directly on the renin-angiotensin system can cause injury and even death to the developing fetus.
36.3.3 Ang II-AT1 Receptor Blockers (ARBs)
Although the prevention of Ang II generation by ACE inhibition is of potential therapeutic benefit, ACE inhibitors may permit an Ang II escape phenomenon. ACE inhibitor-mediated disrupted negative feedback mechanism increases the levels of renin and subsequently of Ang I. In such a situation, non-ACE enzymes such as chymase can convert Ang I to Ang II, resulting in Ang II escape [139]. Therefore, compared to ACE inhibition, selective AT1 receptor blockade might have distinct advantages, like the absence of angiotensin II escape and unopposed AT2 receptor-mediated effects of Ang II, predominantly vasodilation. In addition, the unwanted side effects of ACE inhibitors, such as dry cough and angioedema, are fewer with ARBs.
The primary goal of antihypertensive therapy is reduction in mortality. A meta-analysis of several trials has shown that the use of ACE inhibitors is associated with a 10 % reduction in all-cause mortality. On the other hand, ARBs had a neutral effect on mortality [140]. The lack of superiority in beneficial effects of ARBs (losartan and valsartan) over ACE inhibitors has been attributed to not using a high enough dose of ARBs. In clinical trials where lower doses of ARBs were used, a survival benefit was not found. An insufficient dose of an ARB used in ELITE II, OPTIMAAL, and VALIANT trials might be the reason for an observed lack of beneficial effect. A significant survival benefit in high-risk patients was observed when relatively larger doses of ARBs were used as in LIFE and RENAAL trials [36].
The discovery of drugs targeting AT1 receptors dates back to saralasin, a peptidergic ARB. Because of its short duration of action, partial agonist activity, and low bioavailability, its use was limited to parenteral administration. The breakthrough came with the introduction of losartan, the first orally active non-peptide ARB in 1995, which is about 10,000 times more selective for the AT1 receptor than for the AT2 site. Non-peptide ARBs lack partial agonist activity and are highly specific for antagonizing AT1 receptors linked to the CVS. Structurally, these drugs are classified into three categories, biphenyl imidazole (losartan, valsartan, candesartan, irbesartan, azilsartan, fimasartan), biphenyl carboxylic acid (telmisartan), and imidazole acrylic acid (eprosartan). Fimasartan is available in South Korea [141].
36.3.3.1 Mechanism of Action
All ARBs inhibit Ang II-induced AT1 receptor-mediated vascular contraction, pressor responses, vasopressin release, aldosterone secretion, adrenal catecholamines release and increase in sympathetic tone, and cellular hypertrophy. ARBs are competitive antagonists and possess a high degree of selectivity for the AT1 receptor. The blockade for most of the antagonists except losartan and eprosartan is insurmountable; that is, the maximal response to Ang II cannot be restored in the presence of an ARB. They bind tightly and dissociate slowly, causing the functional loss of occupied receptors. A small difference in the structure and variation in functional groups induces different receptor conformations that result in differences in binding affinity. Thus, losartan has the lowest affinity, whereas irbesartan has the highest affinity among ARBs [reviewed in 142]. Small differences in the molecular structure of each ARB have demonstrated unique molecular effects in experimental studies. Thus, some benefits conferred by ARBs beyond blood pressure control may not be class effects but rather molecular effects as shown in some experimental studies [143]. Some of these effects may account for differences in clinical outcome as shown in Table 36.1 under the column “indication.”
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


