Fig. 49.1
Drugs withdrawn from market between 1990 and 2006 based on overall mechanism of toxicity. The category “QT-Torsades” represents QT interval prolongation and torsade de pointes, while the category “Other” includes aplastic anemia and convulsions. Hepatotoxicity following prolonged administration is listed under “Other” category (Data derived from Shah [4])
Drug-induced long QT syndrome (LQTS, also referred to as acquired long QT syndrome, aLQT) is characterized by QT interval prolongation and increased risk of TdP. In 1964, Selzer and Wray [5] reported QT prolongation and ventricular fibrillation (VF) in response to quinidine. Two years later, Dessertenne [6] described TdP, a polymorphic ventricular tachycardia where QRS complexes “twist” around the isoelectric line in a sinusoidal fashion. Symptoms of TdP include palpitations, syncope, and seizure-like convulsions. TdP is usually self-limited but may degenerate into ventricular fibrillation and sudden cardiac death. A variety of medications from multiple classes have been implicated in drug-induced LQTS and TdP ([7, 8], also see www.crediblemeds.org for an updated listing and also in Chap. 48).
Long QT syndromes linked to mutations of cardiac ion channels have also been identified [9]. Genetic linkage analysis of affected families with congenital LQTS has described several loci associated with LQTS, with most of the affected genes encoding for ion channels or subunits involved in ventricular repolarization (see [6]). The most frequent LQTS variants are loss-of-function mutations in the KCNQ1 (LQT1) and KCNH2 (LQTS2) genes encoding for the slow and rapid components of the delayed rectifier potassium channel currents I Ks (Kv7.1) and I Kr (KV.11.1, also referred to as hERG current). A gain-of-function mutation of the SCN5A gene encoding for the cardiac sodium channel current I Na (Nav1.5) is associated with LQT3. Mutations in genes causing congenital LQTS have been found in patients with drug-induced QT prolongation and ventricular arrhythmia. Congenital LQTS is an inherited disease usually occurring in structurally normal hearts. Beyond genetic evidence, pharmacological evidence exists linking those cardiac ion channels and TdP, and pharmacologic challenge has been used to identify forms of congenital LQTS (see also Chaps. 48 and 52). Most clinically relevant drug-related QT prolongation occurs via inhibition of I Kr, a potassium current mediated in humans by the ion channel KCNH2 (KV11.1) encoded by the human ether-a-go-go-related gene (HERG), analogous to the genetic LQT2 form of the congenital disease (Fig. 49.2).
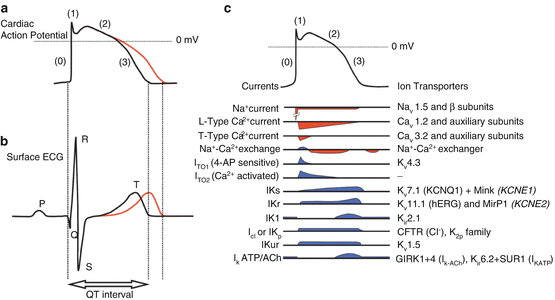
Fig. 49.2
Panels (a, b). Simplified relationship between prolongation of action potential duration and QT interval. Panel (c) illustrates contributions of multiple inward currents (downward deflections, red) and outward currents (upward deflections, blue) to electrogenesis of a generalized ventricular action potential. I Kr/KV11.1 (hERG) and associated subunits MirP1) represent just one of multiple currents functioning during repolarization. I Kur and IKATP/ACh currents are more prominent in atria compared to ventricular myocytes (Adapted with permission from Abriel et al. [8])
49.2 How Does Delayed Repolarization Predispose to TdP and Lead to Proarrhythmia?
Ventricular repolarization is a dynamic process representing a fine balance of inward (depolarizing) and outward (repolarizing) ionic currents. The net outward current that flows with each heartbeat ensures the orderly termination of each cardiac cycle appropriate to the heart rate, modulates cardiac refractoriness and excitability (by defining the time course of reactivation of sodium and calcium currents), as well as affects intracellular calcium handling and cardiac contractility. Drugs that delay repolarization may predispose the myocardium to interruptions in repolarization termed “early after depolarizations” (EADs). EADs represent a form of abnormal electrical activity in which repolarization is interrupted by an inappropriate depolarization that arises during the action potential plateau (or later during the relative refractory period) but prior to full repolarization. EADs can, if of sufficient amplitude and timing, propagate within the ventricles (especially those in which repolarization may be more heterogeneous due to spatiotemporal differences in drug-induced delayed repolarization, creating a proarrhythmic substrate) to elicit premature ventricular excitation that elicit TdP.
It is generally accepted that most drugs that delay cardiac repolarization reduce the rapidly activating delayed rectifier current I Kr encoded by hERG. A historical review of hERG current in cardiac safety studies can be found in Rampe and Brown [10]. Given the multiple components involved in initiating TdP, it is now recognized that drug-induced delayed repolarization is a surrogate marker of TdP proarrhythmia. Indeed, in most instances, drug-induced delayed repolarization simply leads to prolongation of ventricular repolarization (manifest as QT prolongation) that does not result in repolarization instability and triggered activity and is simply reversed upon drug removal. While it is generally accepted that prominent delayed repolarization leads to increased risk for TdP proarrhythmia, the extent of delayed repolarization necessary to elicit EADs or TdP is uncertain and is likely dependent on the presence of covert or overt heart disease and the presence or absence of known risk factors. Such factors include heart rate (bradycardia), electrolyte imbalances (hypokalemia, hypocalcemia, hypomagnesemia), age (older population), sex (female), disease conditions (congestive heart failure, left ventricular hypertrophy, liver function impairment), and congenital long QT syndrome. Many of these risk factors can be considered as reducing “repolarization reserve,” a term that refers to the ability of the myocardium to repolarize in the setting of reduced outward current [11, 12]. Given the integrated response of multiple currents that define repolarization, reducing one outward current may modestly delay repolarization (but not lead to proarrhythmia) due to the remaining multiple currents that support repolarization. However, in the presence of risk factors that may reduce repolarization reserve, additional small reductions in net outward current may have more prominent effects on repolarization stability and predispose to TdP.
49.3 Further Consideration of Drug that Delay Cardiac Repolarization
Emerging data suggest that in most cases, the hERG/I Kr and in vivo QT assays that form the core of the prevailing preclinical ICH S7B Guidance [10] detect drugs that prolong the QT interval in humans. However, there is a growing understanding of instances where “results among preclinical studies are inconsistent and/or results of clinical studies differ from those of preclinical studies” [13]. In these instances, “retrospective evaluation and follow-up preclinical studies can be used to understand the basis for the discrepancies” [13]. Indeed, a comparison of the potency of hERG block of drugs also tested in clinical thorough QT studies revealed a surprising number of false-positive and false-negative findings [14]. Follow-up studies may include studies of drug metabolites and more comprehensive studies of drug effects on multiple cardiac currents (see the CiPA initiative, below). There are drugs (such as verapamil and phenobarbital) that do not prolong the QT interval even though free exposure levels encompass the inhibitory concentration range for prominent outward (repolarizing) I Kr block. In such cases, the lack of concordance is most easily explained by postulating that block of outward (repolarizing) current is mitigated by concomitant block of inward (depolarizing) current during repolarization. Such complex, integrated effects have been called multichannel block [15] or mixed ion channel effects (MICE, [16]). One such candidate current is the l-type calcium current (Cav1.2) that provides depolarizing current that sustains the ventricular action potential (AP) plateau. One can see evidence of multichannel block by changes in the configuration of the AP plateau (representing the integrated activity of multiple ion channels), with calcium channel block reducing repolarization delays elicited by hERG channel block [15]. Alternatively, it is possible to separately evaluate drug effects on multiple cardiac currents (expressed in heterologous systems using higher-throughput automated patch systems; see [17, 18]) and integrate multiple effects using regression techniques [13] or computer reconstructions of ventricular electrical activity [19].
There are instances where a new chemical entity (NCE) is found to block I Kr current and to prolong the QT interval in vivo at plasma concentrations much lower than those expected based on potency of hERG block. In such cases, myocardial drug accumulation may contribute to the reduced safety margin based on hERG IC50 values alone. There is good evidence that this is an issue for some antipsychotic compounds (e.g., risperidone and haloperidol [20]). This combined with the reported ability of hERG to “trap” compounds [17, 21], kinetic considerations that modulate the dynamic block and unblock of I Kr [22], effects of plasma protein binding on drug availability, along with previously mentioned multichannel effects, may provide the basis for pharmacokinetic–pharmacodynamic mismatches that make the preclinical assessment risk of proarrhythmia difficult, even for hERG-selective compounds. In addition, there is evidence that some compounds indirectly reduce hERG current by reducing trafficking and/or expression of the channel protein to the sarcolemma with chronic exposure [20]. For such a mechanism, simple assays have be devised to measure the change in surface channel expression [23, 24]. Inhibition of hERG transcription, for example, by desethylamiodarone, a metabolite of amiodarone, has been suggested as a possible mechanism to explain the delayed QT-prolonging effects of amiodarone [25].
Drugs that affect autonomic tone may also lead to conflicting data: the complex relationship between QT and RR intervals may change depending on prevailing autonomic tone [26]. If species-specific, such effects could show no drug response preclinically but result in QT effects clinically (even using individual QT–RR plots to derive corrected QT values).
49.4 Preclinical Assays to Detect Delayed Repolarization
49.4.1 In Silico
Computational models have been developed to predict the hERG potency of NCEs (see [27]). While useful in early screening campaigns and guiding chemical synthesis, these models are not widely accepted for subsequent proarrhythmic risk assessments.
49.4.2 In Vitro
Owing to the emphasis on hERG current in regard to delayed proarrhythmia, numerous approaches have been used to evaluate drug effects on hERG current. In general, these approaches may be categorized as direct or indirect, based on the parameter(s) evaluated. Direct (functional) assessments provide determinations of IC50 values for block of hERG current expressed in heterologous expression systems measured using voltage clamp techniques. While formerly assessed using manual whole cell voltage clamp techniques, such studies have largely been supplanted by automated patch clamp platforms (see [18]). Examples of a less direct assessment include studies involving the measurement of Rb+ or Thallium+ flux with HEK or CHO cells transfected with I Kr channels and binding assays which measure displacement of potent hERG ligands such as radiolabeled dofetilide, MK-49, and astemizole. Indirect measures are more useful in detecting potential hazards associated with hERG, such as screening larger numbers of drug candidates prior to lead selection. Indirect measures are more difficult to translate into clinical risk assessments (typically as “safety margins”), which are typically based comparisons of IC50 values for hERG block vs. clinical exposures (based on C max values) with considerations for plasma protein binding.
49.4.3 Proarrhythmic Models
There is a tacit assumption in ICH S7B that the integrated risk assessment for QT interval prolongation will reflect the risk of TdP. However, since QT interval prolongation is an imperfect surrogate marker of proarrhythmia, and the incidence of TdP is so low as to require inappropriately large numbers of patients in clinical studies to define the proarrhythmic risk, potentially useful in vitro and in vivo proarrhythmia models have been developed and described [28–30].
Since the identification of TdP, proarrhythmic risk has been investigated using mechanistic studies of inherited LQTS and proarrhythmic properties of several antiarrhythmic drugs (see Bialecki et al. [31], Chap. 48). A key element present in many such models is the need for risk factors linked to TdP clinically, such as female gender, bradycardia, hypokalemia, or autonomic imbalance. They are therefore deemed “sensitized” models and are used principally as follow-up studies to confirm potential risk factors or in parallel to drug development when a family class effect is expected. The mechanisms of TdP have been well investigated, and evidence suggests that rhythm instability (such as rapid transitions from long to short cardiac cycle lengths) is often observed in adults just prior to TdP initiation [32–34]. The enhanced sensitivity likely increases spatial and temporal repolarization heterogeneity, enhancing the proarrhythmic substrate [35]. Despite such information, the QT interval duration remains as a popular (yet imperfect) surrogate. Alternative surrogates have included EADs, ectopic beats, dispersion of ventricular repolarization [36], heterogeneity and hysteresis of restitution [37], electrical alternans (action potential duration alternans) [38], beat-to-beat instability of QT interval duration [37], and electromechanical window [39]. In the absence of ventricular arrhythmias, it is not possible to define a risk threshold and, therefore, fully establish the predictive value of these surrogates. The comprehensive in vitro proarrhythmia assay initiative (CiPA, described below) represents an evolving preclinical paradigm that focuses on linking recognized proarrhythmic mechanisms of drugs with clinically adjudicated proarrhythmic risk.
49.4.4 In Vitro SCREENIT™ Rabbit Heart
This isolated heart preparation developed by Hondeghem (SCREENIT™ [40]) evaluates pro- or antiarrhythmic properties of NCEs using retrogradely perfused isolated female rabbit heart preparations according to the classical Langendorff technique. In this model, bradycardia is induced by AV node ablation and the heart is alternately paced at fast and slow stimulation rates; additional sensitization is accomplished using a hypokalemic buffer. Extracellular monophasic action potential recordings and ECG recordings may be used to evaluate proarrhythmic risk; triangulation of the monophasic action potential duration, reverse use dependence (greater prolongation of the repolarization at slow vs. rapid stimulation rates), and beat-to-beat variability of repolarization (temporal instability) are three major components of proarrhythmic risk in this model (leading to the acronym of TRIaD; for reviews, see Lawrence et al. [28] and Thomsen et al. [29]). This model, validated in a blinded assay on additional sets of positive and negative reference compounds, was shown to detect QT positive compounds and, more interestingly, selective in excluding false positives. Furthermore, the use of TRIaD parameters allows identification of those NCEs for which monophasic action potential duration prolongation is linked to antiarrhythmic activity.
49.4.5 The In Vitro Wedge Preparation
This preparation is also useful to demonstrate the effects of NCEs on repolarization variability (i.e., heterogeneity). Following the demonstration of transmural heterogeneity in guinea pig papillary muscles [41], it was specifically designed to measure transmural dispersion across the ventricular wall [42, 43] with a special emphasis on M cells (localized in the midmyocardium of in vitro preparations) that were demonstrated to be very sensitive to AP prolongation by drugs and risk factors for TdP. In this technically difficult assay, drugs are perfused through native coronary arteries, and transmembrane action potentials are simultaneously recorded from epicardial, midmyocardial, and endocardial layers (obtained from edges of the sliced wedge), while an ECG-like trace is obtained across the preparation (typically rabbit hearts). The proarrhythmic surrogates used in this model include APD (specifically those from midmyocardial cells), and the interval between the peak and end of the pseudo T wave (QTpeak–QTend) is used as an index of transmural dispersion of repolarization (TDR, [44]). Indices of TDR may be useful to quantify proarrhythmic risk [40]. This model demonstrates the proarrhythmic potential of multiple proarrhythmic drugs [45–47] consistent with clinical outcomes. However, it should be noted that the existence of M cells in intact human hearts is still a matter of debate and has not been unequivocally demonstrated [48, 49].
49.4.6 In Vivo Chronic Atrioventricular (AV) Block
Based on bradycardia and electrolyte disorders identified as prominent risk factors for proarrhythmia, a chronic AV block dog model was developed and extensively used to analyze the mechanisms and risk factors associated with the use of Class I and Class III antiarrhythmic drugs, either alone or in combination (see [29, 31]). In this model, bradycardia is induced by formaldehyde injection into the AV node, resulting in chronic AV block; ventricular hypertrophy, electrical remodeling and rhythm instability that result all increase the susceptibility to TdP, while additional predisposing factors (hypokalemia) may further sensitize the model. The model may be modified to promote TdP with specific pacing algorithms to mimic the typical short–long–short sequences linked to the initiation of TdP and used to test pharmacological compound classes such as cisapride, almokalant, sertindole, or astemizole. In this model, QT or MAPD prolongation has been reported as a poor predictor of TdP, and other markers such as beat-to-beat variability seem to be more appropriate. Due to its very low throughput and technical challenges, this model has not been validated against a large panel of drugs, although it has been described as reproducible and highly sensitive for testing arrhythmogenic properties of drugs. A comparable model using Cynomolgus monkeys has also been developed (see [50]).
49.4.7 In Vivo Methoxamine-Sensitized Rabbit
In this in vivo model, chloralose-anesthetized rabbits treated with methoxamine (an α1 adrenoceptor agonist) develop an increased sensitivity to delayed repolarization due to improper calcium handling leading to induction of ectopic beats [51, 52]. These effects are specifically marked for I Kr blockers, leading to prominent QT prolongation, U waves, and finally polymorphic ventricular tachycardia. While arrhythmias in this model are not associated with bradycardia, the electrophysiologic manifestation is similar to clinical TdP. Apart from the typical Class III antiarrhythmic compounds [53], this model has been used for several other NCEs including antibiotics such as sparfloxacin, levofloxacin, gatifloxacin, and Prulifloxacin [54, 55]. Interestingly, the proarrhythmic drug quinidine is ineffective in inducing arrhythmias in this model, probably because of its α1-blocking properties; this model may also be insensitive to terfenadine [56], unless high doses are used [57]. The sensitivity to drugs other than Class III antiarrhythmic compounds remains to be demonstrated and limits the utility of this model in preclinical safety assessment.
49.5 Translation of Preclinical to Clinical Findings
In most cases, hard evidence regarding the predictive value of electrophysiologic preclinical testing is not readily available in the public domain. With a negative preclinical finding, subsequent development of a drug candidate may be halted and limited (if any) information provided in the public domain. Consequently, the scientific and medical community is left with high-profile examples of side effects in humans apparently not detected during preclinical assessments. Data have been generated to assess the value of preclinical tests to predict the potential of NCEs to prolong the QT interval of the ECG (and presumably the proarrhythmic potential of these drugs).
Published data suggesting that a 30-fold margin between the highest free C max of a drug in clinical use and the hERG IC50 could be adequate to ensure an acceptable degree of safety from arrhythmogenesis with a low risk of obtaining false positives [58–60]. Published literature looking at the predictive value of preclinical QT-related models to humans is emerging [61–63]. Further work is required to extend the validation set in order to determine the confidence in the models in terms of their capacity to mimic the same physiological/pathophysiological mechanisms in humans and the confidence in their translation to humans (i.e., sensitivity, specificity, predictive value, as well as prevalence [1, 14, 16, 61]). However, there are areas that should be carefully considered to ensure optimization of the assays and ultimately increase the predictive value of preclinical testing. These include but are not limited to (1) species differences in the expression or functionality of the molecular target mediating the adverse effects, (2) differences in PK properties between test species and humans, (3) sensitivity of the test system, (4) optimization of the test conditions, (5) appropriate statistically powered study designs, (6) appropriate timing of functional measurements in relation to the time of maximal effect, (7) delayed/chronic effects of parent drug and/or metabolites, (8) difficulty of detection in animals using standard studies (e.g., arrhythmia), and (9) assessment of a suboptimal surrogate end point that predicts with some degree of confidence the clinical outcome (e.g., QT/QTc interval prolongation as a surrogate of TdP).
49.6 Clinical Considerations: QT Prolongation
This ICH E14 guidance issued in 2005 established the so-called clinical thorough QT (TQT) study whose focus was “to assess the potential of a drug to delay cardiac repolarization” [64]. TQT studies are designed to exclude a “threshold effect” below which QTc prolongation is considered to have no significant clinical consequence. This threshold effect is interpreted as a placebo-corrected change from baseline QTc (double-delta QTc) demonstrated by the two-sided 90 % confidence interval. A negative TQT study is concluded with an effect size of about 5 ms or less with the upper bound of the two-sided 90 % confidence interval below 10 ms. When this limit is exceeded (defining a “positive” TQT study), the QTc effect may significantly exceed 5 ms, and the upper bound of the two-sided 90 % confidence interval may exceed 10 ms. This definition was chosen to provide reasonable assurance that the mean drug effect on the QTc interval for therapeutic and reasonable supratherapeutic exposures is not greater than around 5 ms. In instances of a positive TQT study, further studies with intensive ECG monitoring in later stage clinical trials are encouraged to assess proarrhythmic risk. TQT studies typically include a negative (placebo) control for circadian variation, a positive control (typically moxifloxacin administered orally as a 400 mg single oral dose), and at least one dose of drug candidate expected to result in plasma concentrations that includes the maximum systemic exposure anticipated in the target population. While typically conducted in normal volunteers, less stringent evaluations may be conducted for oncologic drugs in patients with cancer (e.g., with cytotoxic agents [65]).
Human TQT studies are resource intensive requiring attention to specific details to ensure the necessary rigor to quantify small changes in the QTc interval (5 ms resolution vs. baseline values in the range of 400–450 ms, i.e., 1–2 %). The reliable and consistent assessment of the end of the QT interval was an earlier challenge that has been largely resolved by the use of qualified computer algorithms. Another challenge was posed by the fact that the QT interval varies with heart rate, with longer QT intervals at slower rhythms that may confound a drug’s direct effect on repolarization. Presently, correction approaches employed include fixed formulae (Fridericia’s formula, considered more appropriate compared to the previously popular Bazett’s correction [66–68]), individualized correction factors (corrections based on RR–QT interval relations established for each subject), or beat-to beat measures [69, 70]; optimal fixed heart rate correction formulae also vary across species [71].
49.7 Clinical Practice Perspectives
The drugs frequently used in clinical practice that carry the greatest risk for QT prolongation and TdP are Class III (and some older Class I) antiarrhythmic drugs (such as sotalol, dofetilide, ibutilide, and azimilide) for which hERG inhibition and QT prolongation are part of the therapeutic mechanism of action; the observed rate of TdP among these patients is about 1 % [72] (Chaps. 48 and 52). It is also likely that cardiac pathologies in this patient population predispose to proarrhythmic risk [73]. In addition, the presence of predisposing factors including hypomagnesemia, hypokalemia, bradyarrhythmias, coronary artery disease, and pre-existing cardiac rhythm disturbances may contribute to the incidence of TdP.
Noncardiac drugs can also cause TdP, such as antibiotics, antipsychotics, and antidepressants, for which TdP or QT prolongation is an undesirable side effect of the drug. The risk of TdP is lower than with Class III antiarrhythmic drugs (ranging from 0.01 to 0.1 % [74, 75]); however, these drugs are prescribed more frequently and with far less awareness for monitoring of the QT effects than the class III antiarrhythmic drugs [76]. Examples of drugs which have to be taken off the market due to the development of TdP include the gastrointestinal drug cisapride (Propulsid) and second-generation antihistamines astemizole (Hismanal) and terfenadine (Seldane; see [4]). For some drugs, warnings of the risk of QT prolongation and TdP were added to labeling, for example, droperidol (Inapsine).
TdP in clinical practice is often associated with polypharmacy and drug–drug interactions. A common pharmacodynamic interaction includes inadvertent prescription of multiple agents with hERG-blocking properties to a patient. For example, a patient taking a stable dose of a Class III antiarrhythmic agent for atrial fibrillation may present with an acute infection to a general practitioner in a primary care clinic or urgent care facility who is unaware of the situation and may prescribe a quinolone antibiotic that may predispose to TdP. In a review of medication records of >1 million patients, prescriptions of 1 QT-prolonging medication was identified in 23 % of patients, two agents in 9 % of patients, and three agents in 0.7 % of patients [77]. While these interactions rarely cause adverse outcomes, the concomitant use of multiple contraindicated drugs may cause drug-induced TdP in a substantial proportion of patients.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


