Fig. 38.1
Schematic of the nephron illustrating the handling of water and electrolytes by the different segments and the major nephron sites of diuretic action. Aldosterone effect influences sodium and potassium handling. Heavy arrows represent the approximate percentage of sodium reabsorbed by the various nephron segments
Table 38.1
Pharmacokinetics of diuretics
Half-life | ||||
|---|---|---|---|---|
Diuretic | Bioavailability (%) | Normal subjects (hrs) | Renal failure (hrs) | HF (hrs) |
Loop of Henle | ||||
Furosemide | 10–>100 | 1.5–2 | 2.8 | 2.7 |
Bumetanide | 80–100 | 1 | 1.6 | 1.3 |
Torsemide | 80–100 | 3–4 | 4–5 | 6 |
Ethacrynic acid | 90–100 | 1–3 | ND | ND |
Distal convoluted tubule | ||||
Bendroflumethiazide | ND | 5–9 | ND | ND |
Chlorthalidone | 64 | 50–60 | ND | ND |
Chlorothiazide | 30–50 | 1.5–2.5 | ND | ND |
Hydrochlorothiazide | 60–80 | 3.2–13.1 | Increased | ND |
Indapamide | 93 | 15–25 | ND | ND |
Metolazone | 40–60 | 15 | NDa | NDa |
Polythiazide | ND | 26 | ND | ND |
Trichlormethiazide | ND | 1–4 | 5–10 | ND |
Distal/collecting duct | ||||
Amiloride | 50–60 | 17–26 | 100 | ND |
Triamterene | >80 | 2–5 | Prolonged | ND |
Spironolactone | b | 1.5 | ND | ND |
Eplerenone | 70 | 4–6 | Small↑ | NDc |
38.3 Carbonic Anhydrase Inhibitors
Acetazolamide is the only carbonic anhydrase inhibitor with relevant diuretic effects. Acetazolamide binds tightly to carbonic anhydrase, and high concentrations are found in tissues containing this enzyme, particularly red blood cells and the renal cortex. Acetazolamide is mainly renally cleared as the unchanged molecule with approximately 50 % of its renal clearance as the result of active tubular secretion. Its administration is ordinarily accompanied by a brisk alkaline diuresis. Although carbonic anhydrase inhibitors are proximal tubular diuretics (where the bulk of Na+ reabsorption occurs), their net diuretic effect is modest, since Na+ reabsorption in more distal nephron segments offsets proximal Na+ losses. Acetazolamide use is constrained by both its transient action and the development of metabolic acidosis with prolonged use. Acetazolamide (250–500 mg daily) can correct the metabolic alkalosis that derives from chloride (Cl−) loss with thiazide and/or loop diuretic therapy and can be of some utility in patients with sleep apnea [5, 6].
38.4 Loop Diuretics
Loop diuretics act predominately at the apical membrane in the thick ascending limb (TAL) of the loop of Henle, where they compete with Cl− for binding to the Na+/K+/2Cl− cotransporter, thereby inhibiting Na+ and Cl− reabsorption [7]. Loop diuretics also have effects on Na+ reabsorption within other nephron segments, which are qualitatively minor compared with their action at the TAL. Other clinically important effects of loop diuretics include a decrease in both free water (H2O) excretion and absorption during H2O loading and dehydration, respectively, about a 30 % increase in fractional calcium (Ca++) excretion, a significant increase in magnesium (Mg++) excretion, and a transient increase followed by an ultimate decrease in uric acid excretion [8]. The increase in urine Ca++ excretion with loop diuretic therapy increases parathyroid hormone release and therein a negative cardiac effect relating to the profibrotic effect of PTH on the myocardium [9].
The available loop diuretics include bumetanide, ethacrynic acid, furosemide, and torsemide. These compounds are heavily protein bound (to albumin), and therefore, in order to gain access to the tubular lumen (site of action), they must undergo secretion (the same applies to thiazide-type diuretics), which in their case is by way of probenecid-sensitive organic anion transporters localized to the proximal tubule [10, 11]. Tubular secretion of loop diuretics may be reduced in the company of elevated levels of endogenous organic acids, such as in chronic kidney disease (CKD), and by drugs that share the same transporter, such as salicylates and nonsteroidal anti-inflammatory drugs (NSAIDs). Loop diuretic protein binding can also be decreased by uremic toxins and/or fatty acids, which can in kind alter diuretic action.
Diuretic excretion rates approximate drug delivery to the medullary TAL and match up well with the observed natriuretic response [2, 4]. The relationship between urinary loop diuretic excretion rate and natriuretic effect is that of an S-shaped sigmoidal curve (Fig. 38.2) [11]. A normal dose-response relationship (as is typically seen in the untreated patient with hypertension) can be distorted (downward and rightward shifted) by a mixture of clinical conditions ranging from volume depletion (“braking phenomenon”) to HF or nephrotic syndrome (disease-state alterations) to various drug therapies. As an example of the latter, NSAIDs modify this dose-response relationship by inhibiting prostaglandin synthesis, blunting any expected diuretic effect. Finally, although the diuretic dose-response relationship can flatten in the setting of nephrotic-range proteinuria, the binding of loop diuretics to urinary protein and thereby limiting free drug access to receptor sites seems not to be the basis for this reduction in diuretic effect [12].
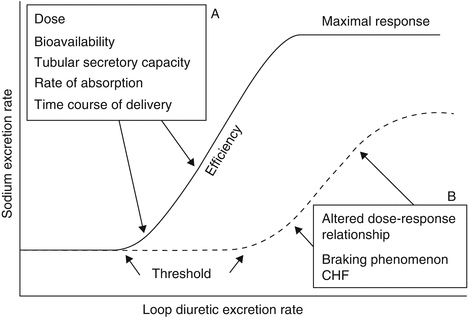
Fig. 38.2
Pharmacokinetic (A) and pharmacodynamic (B) determinants of the natriuretic response to a loop diuretic. The broken line represents a distorted dose-response relationship, as is seen in a patient with diuretic resistance. The amount of diuretic needed to achieve a threshold response can vary quite substantially in the presence of diuretic resistance
38.4.1 Furosemide
Furosemide is the most broadly used diuretic in this class; however, its use is made more difficult by erratic absorption with a bioavailability range from 12 to 112 % [13]. The absorption coefficient of variation varies from 25 to 43 % for different furosemide products; thus, exchanging one furosemide formulation for another will not automatically standardize patient absorption and thus response, when furosemide is given orally. Bumetanide and even more so torsemide are more predictably absorbed than is furosemide. The consistency of torsemide’s absorption and its longer duration of action are pharmacologic features to consider when loop diuretic therapy is necessary in the HF patient [14].
38.4.2 Bumetanide/Torsemide
The loop diuretics furosemide, bumetanide, and torsemide are diuretics of choice in the patient with CKD [15]. The pharmacokinetics of loop diuretics are altered in the CKD population: with renal clearance of these drugs being reduced corresponding to the degree of change in renal function. In general, furosemide’s pharmacokinetics are more significantly altered in CKD than is the case for other loop diuretics, since furosemide is a renally glucuronidated compound [16]; therefore, both its renal metabolic and intact clearances are dually reduced in CKD. Alternatively, bumetanide and torsemide undergo significant hepatic metabolism; thus, their pharmacokinetic profiles in CKD vary only as the result of decreased renal clearance of the intact molecules (Table 38.1).
38.4.3 Ethacrynic Acid
Ethacrynic acid is structurally different from other loop diuretics. Its diuretic effect is similar to that of other loop diuretics. Ethacrynic acid has little or no effect on glomerular filtration or on renal blood flow, except following pronounced reductions in plasma volume when associated with rapid diuresis. It is used much less frequently than the other loop diuretics mainly because of an increased propensity to hearing changes particularly when given intravenously [17]. Its main use clinically is in those patients with documented sulfa allergies in that it is the only loop diuretic that is structurally different in not having a sulfonamide molecule [18].
38.5 Adaptation to Diuretic Therapy
Diuretic-induced inhibition of Na+ reabsorption in one nephron segment elicits important adaptations in other nephron segments, which not only limits diuretics’ antihypertensive and fluid-depleting actions but also contributes to the magnitude of side effects. Although a portion of this resistance to diuretic effect is an expected consequence of their use, profound diuretic resistance from such adaptations can be encountered in patients with clinical disorders such as HF, cirrhosis, and/or proteinuric CKD. An understanding of how adaptation to diuretic therapy occurs is a prerequisite to minimizing the negative features of this process.
The initial dose of a diuretic ordinarily produces a brisk diuresis and in most instances results in a net negative Na+ balance. The new equilibrium state established with diuretic therapy is one where body weight decreases and with repetitive dosing stabilizes, since adaptive processes intervene and preclude a continuous volume loss. In nonedematous patients given either a thiazide or a loop diuretic, this adaptation or braking phenomenon occurs within a matter of days and limits weight loss to 1–2 kg; this has been demonstrated in normal subjects given the loop diuretics furosemide or bumetanide. Furosemide administered to subjects ingesting a high-Na+ diet (270 mmol/24 h) produces a brisk natriuresis, which results in a negative Na+ balance for the following 6 h. This is followed by a prolonged period of time up until the next dose, where Na+ excretion is reduced to a level considerably below that of intake [19]. This post-diuresis Na+ retention compensates for initial Na+ losses, with the result being no net weight loss. In fact, this same pattern of Na+ loss and compensatory retention can persist for as long as a month after furosemide administration [20]. Sodium intake, prior to and after diuretic dosing, will influence the end results of the braking phenomenon. For example, if Na+ intake is kept low, Na+ balance will remain negative in the hours after the initial natriuresis with a net fall in body weight.
38.6 Mechanisms and Management of Diuretic Resistance
Diuretic resistance is encountered in a number of disease states, such as CKD, nephrotic syndrome, HF, and cirrhosis with ascites, and most times relates to prominent pharmacodynamic alterations that are derived from disease-state organ dysfunction [11]. Combination diuretic use, which produces sequential nephron segment blockade and thus an additive diuretic response, is frequently necessary and is regularly employed in these conditions. Pharmacokinetic determinants of diuretic response, including the dose administered, absolute bioavailability, and tubular transport rate/capacity are pertinent considerations factors in how a diuretic response is generated [11]. Pharmacodynamic factors, such as improvement in the underlying disease-state condition promoting diuretic resistance, are perhaps more important to overall response and often result in modification of the dose-response relationship. Stratagems used to maximize the diuretic response to loop diuretics include correcting abnormal hemodynamic parameters, utilizing larger doses of well-absorbed orally administered loop diuretics, or using constant intravenous infusions or bolus diuretic therapy [21]. Bolus loop diuretic therapy does not appear to be much different than that of loop diuretic infusions in initiating and maintaining a diuretic response [22]. If these measures fail, then diuretic combinations are useful. Perhaps the most effective is the combination of metolazone and a loop diuretic. The rationale for and use of various diuretic combinations, with particular emphasis on the metolazone-loop diuretic combination, is well established but requires a deft touch in selection of medication doses in that over-diuresis is not uncommon [23].
38.7 Thiazide Diuretics
The main site of action for thiazide-type diuretics is the early distal convoluted tubule where the coupled reabsorption of Na+ and Cl− is inhibited (Fig. 38.1). Besides effects on Na+ excretion, thiazide diuretics also impair urinary diluting capacity (while preserving urinary concentrating mechanisms), reduce Ca ++ and uric acid excretion, and are modestly magnesuric. The latter can be particularly prominent with long-acting thiazide-type diuretics, such as chlorthalidone [24].
38.7.1 Hydrochlorothiazide
Hydrochlorothiazide (HCTZ) is the most widely used thiazide-type diuretic in the United States. Its absorption is dose proportional with a bioavailability ranging from 60 to 80 %. The rapidity and extent of its absorption can be reduced in HF and/or CKD. The onset of diuresis with HCTZ is fairly rapid occurring within 2 h and peaking between 3 and 6 h and sometimes continuing for as long as 12 h; however, only a small fraction of the total natriuretic response to HCTZ occurs beyond 6 h after dosing. The diuretic effect of HCTZ can be extended by administering doses in the 50–200 mg range [25]. Alternatively, if a more extended natriuresis is sought after, a longer-acting thiazide-type diuretic – such as chlorthalidone or metolazone – can be considered.
The half-life of HCTZ and other thiazide diuretics is extended in patients with HF and/or CKD and is particularly lengthening when these disease states are decompensated. Doses of HCTZ in the order of 100–200 mg/day can induce diuresis in patients with CKD, which is contrary to the belief that these drugs are ineffective in advanced stages of CKD [25, 26]. However, in CKD the magnitude of the natriuretic response to a thiazide diuretic has a ceiling controlled by two factors: first, the lowered GFR in CKD reduces the absolute filtered load of Na+, and second, the distal tubular site of action for a thiazide diuretic is a locus where even under the best of circumstances, only a modest natriuretic response can be anticipated [25, 26].
38.7.2 Metolazone
Metolazone is a quinazoline diuretic with a predominant distal tubular site of action and a minor inhibitory effect on proximal Na+ reabsorption through a carbonic anhydrase-independent mechanism. Metolazone is lipid soluble and has a large volume of distribution (V D), which plays a role in its extended duration of action. The pharmacokinetic features of metolazone are a factor in its effectiveness in the setting of either CKD or diuretic-resistant situations, particularly when combined with a loop diuretic [11, 24]. Oral metolazone in its Zaroxolyn form is slowly and rather erratically absorbed. Diuretic resistance, the failure to respond to a diuretic regimen, is usually taken to signify a worsening of the primary volume-retaining state, but with metolazone, it simply can be a consequence of limited drug absorption [11, 24].
38.7.3 Chlorthalidone
Although chlorthalidone and HCTZ are structurally similar compounds, they are very dissimilar pharmacokinetically. Chlorthalidone is distinguished from HCTZ in having an extremely long half-life and a very large volume of distribution owing to its extensive partitioning into red blood cells [27]. This latter feature creates a substantial depot for chlorthalidone, allowing for a slow streaming effect (red cell → plasma) with subsequent gradual elimination from the plasma compartment by tubular secretion. The extremely long half-life of 40–60 h for chlorthalidone differentiates it from HCTZ, which has a much shorter but wider variation in half-life, from 3.2 to 13.1 h [27]. This plasma half-life difference can be expected to correlate with a more extended effect of chlorthalidone on diuresis and possibly BP. Moreover, the post-diuretic period of antinatriuresis, otherwise termed the “braking phenomenon,” is less apt to interfere with net Na+ loss when a long-acting diuretic, such as chlorthalidone, is administered. Also, chlorthalidone being present in the blood for a longer period of time lengthens more drug exposure in tissue compartments where the drug has its putative effects; however, diuretic blood levels needed to effect direct vessel dilation per se are typically several times higher than what is achieved therapeutically; therefore, it is unclear how the blood level of a diuretic, such as chlorthalidone, might relate to a direct vascular mechanism of action [28, 29].
38.7.4 Indapamide
Indapamide is the first of a class of antihypertensive diuretic agents known as indolines, and it is a mildly natriuretic antihypertensive agent. Indapamide has excellent and almost complete bioavailability, and its metabolism follows linear kinetics with a half-life of ≥15 h. There is little accumulation of indapamide in renal failure presumably because of a shift to greater biliary elimination [30]. At doses of 5 mg or more/day, the diuretic effect of indapamide can be substantial. At 2.5 mg/day fractional Na+ excretion is unchanged when measured after 4 weeks of treatment; thus, at the usual therapeutic doses of 1.25–2.5 mg/day, the antihypertensive effect of indapamide seems to be related as much to calcium antagonist vasorelaxant effects as to its diuretic property, which has been suggested with its use in end-stage renal disease (ESRD) [31]. Like several other thiazide-type diuretics, indapamide affords the treated patient with hypertension cardiovascular (CVR) risk reduction in the setting of diabetes, left ventricular hypertrophy, and/or stroke [32].
38.8 Distal Potassium-Sparing Diuretics
There are two classes of K+-sparing diuretics: competitive antagonists of aldosterone, such as spironolactone and eplerenone, and compounds – such as amiloride and triamterene – which work in an aldosterone-independent manner. Drugs in this class inhibit active Na+ absorption in the late distal tubule and the collecting duct. In so doing, basolateral Na+– and K+-ATPase activity drops and intracellular K+ concentration is reduced. The resultant decrease in the electrochemical gradient for both K+ and H+ reduces secretion of these cations. Potassium-sparing diuretics also reduce Ca++ and Mg++ excretion and can be of some utility in patients prone to the development of calcium-containing stones [33]. Since K+-sparing diuretics are only modestly natriuretic, their clinical utility resides more in their K+-sparing properties, especially when more proximally acting diuretics increase distal Na+ delivery, or in states of primary aldosteronism. Potassium-sparing diuretics do, however, have a significant natriuretic effect when they are one of several components of a diuretic regimen, so-called sequential nephron blockade, and/or when used in suitably titrated doses in patients with cirrhosis/ascites [34].
38.8.1 Spironolactone
Spironolactone is a highly protein-bound and well-absorbed, lipid-soluble K+-sparing diuretic. 7α-Thiomethylspirolactone and canrenone are two metabolites of spironolactone, which are responsible for much of its antimineralocorticoid activity [35]. The onset of action for spironolactone is characteristically slow, with a peak response at times 48 h or more after the initial dose. Currently, spironolactone is used with increasing regularity in patients with resistant hypertension (Chap. 42), with or without primary aldosteronism, who are receiving multidrug regimens that include a diuretic and any other of the several drug classes, including an angiotensin-converting enzyme (ACE) inhibitor, angiotensin II-AT1 receptor blocker (ARB), and/or a calcium channel blocker (CCB) [36]. Neurohumoral screening with high plasma aldosterone values and suppressed plasma renin activity values are markers for likelihood of a therapeutic response but not the magnitude of response per se; however, the response to spironolactone can still be significant even without fully suppressed plasma renin activity values and/or elevated plasma aldosterone values. Spironolactone also affords mortality and morbidity benefits in patients with HF and a reduced ejection fraction [37]. Spironolactone remains active in states of reduced renal function because it gains access to its site(s) of action independent of glomerular filtration. The propensity of spironolactone to cause hyperkalemia precludes its use in many HF and/or CKD patients; however, this appears to be less of an issue in patients on maintenance hemodialysis even in the setting of functional anuria [38].
38.8.2 Eplerenone
Eplerenone is a mineralocorticoid receptor antagonist with a molecular structure that affords greater selectivity for the aldosterone receptor; accordingly, its reduced affinity for androgen and progesterone receptors results in less gynecomastia than is seen with spironolactone [39]. Unlike spironolactone, eplerenone does not have any active metabolites, and it is broken down by CYP3A4; thus, coadministration of CYP3A4 inhibitors, such as verapamil and diltiazem, will result in moderate increases in plasma levels of eplerenone. Typically, eplerenone is a very mild diuretic; thus, its antihypertensive effects originate from non-diuretic aspects of its action. Such actions result in a level of BP reduction comparable to that seen with drug classes such as ACE inhibitors (Chap. 36) and CCBs (Chap. 37) [40, 41]. Eplerenone therapy also results in regression of left ventricular hypertrophy (LVH) (either when given alone or when administered with an ACE inhibitor) as well as a prominent anti-proteinuric effect as is similarly observed with spironolactone [42, 43]. In patients with a recent history of acute myocardial infarction (MI) and left ventricular dysfunction or HF, eplerenone reduces morbidity and mortality when added to the pharmacologic standard of care [44].
38.8.3 Amiloride
Amiloride is a K+-sparing diuretic, which is actively secreted by cationic transporters present in the proximal tubule. Amiloride blocks epithelial Na+ channels (ENaC) in the luminal membrane of the collecting duct, but only a modest natriuretic response can be expected with its use. Amiloride has but a modest antihypertensive effect when given as monotherapy. Its BP-lowering effect can be considerably enhanced when coadministered with a thiazide diuretic [45]. Unlike HCTZ, amiloride appears not to have a negative effect on glucose homeostasis [46]. Amiloride has a longer half-life than triamterene and, thus, can be administered less frequently than triamterene to utilize its ability to preserve potassium balance in the diuretic-treated patient [47]. Amiloride is extensively renally cleared and will accumulate with its repetitive dosing in the setting of reduced renal function. If amiloride therapy is needed in the setting of CKD (GFR <50 mL/min), either the dose should be reduced or the dosing frequency decreased to lessen the likelihood of developing hyperkalemia.
38.8.4 Triamterene
Triamterene is another K+-sparing diuretic, which works independently of a direct antagonism of aldosterone effects. Triamterene is metabolized to an active phase II sulfate-conjugated metabolite. Both triamterene and its sulfated metabolite are cations and gain access to their intraluminal site of action by proximal tubular secretion. Triamterene and its active metabolite accumulate upon repetitive dosing in the patient with CKD. In those unusual circumstances where its use is considered necessary in CKD, empiric dosage adjustment is advisable because of the potential for developing hyperkalemia. Because of its weak BP-lowering properties, triamterene is seldom employed as monotherapy for hypertension. It is usually used in combination with a thiazide-type diuretic with the premise behind such a two-diuretic combination being that triamterene reduces the K+ and Mg++ losses that might otherwise accompany thiazide therapy [48]. Triamterene, given together with a NSAID, has been reported to cause acute kidney injury, which may last for several days. The mechanism behind this interaction is unclear but may relate to triamterene’s increasing renal vascular resistance (and as much as a 30 % decrease in RBF) – correspondingly, there is an increase in the urinary excretion of the vasodilator prostaglandins E2 and F2 [49]. The decrease in prostaglandin production, which follows NSAID therapy, would then allow for an exaggerated renal vasoconstrictor effect from triamterene [49].
38.9 Mechanism of Diuretic Action
The effect of a thiazide diuretic on BP may be divided into three successive phases: acute, subacute, and chronic, which correspond to periods of about 1–2 weeks, several weeks, and several months, respectively [51]. In the “acute” response phase, the BP-lowering effect of a diuretic is coupled to a reduction in ECF volume and a corresponding decrease in cardiac output. The early response (first 2–4 days of treatment) to a thiazide-type diuretic, in the setting of a “no-salt-added” diet (100–150 mmol/day), results in a net Na+ loss ranging from 100 to 300 mmol, which translates into a 1–2 L decrease in ECF volume. This decrease in plasma volume reduces venous return and diminishes cardiac output, the basis for the initial BP decrease with a thiazide diuretic. In due course the thiazide diuretic effects on volume and cardiac output lessen in importance, although BP remains lowered. During the subacute phase of a treatment response (first few weeks), plasma volume returns to slightly less than pretreatment levels, despite the continued administration of a diuretic. The subacute response phase with thiazide-type diuretics is a transitional period during which both volume and resistance factors both contribute to the observed BP reduction (Fig. 38.3).
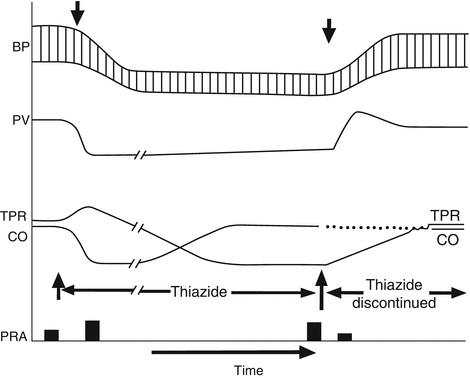
Fig. 38.3
Effects of thiazide administration in an “idealized” patient with hypertension. Early reductions in plasma volume are gradually corrected and supplanted by peripheral vasodilation as the primary BP-lowering effect of these drugs. BP blood pressure, CO cardiac output, PRA plasma renin activity, PV plasma volume, TPR total peripheral resistance
In the chronic response phase of therapy, the BP reduction with a thiazide diuretic reverts to that of a reduction in total peripheral resistance (TPR). The decrease in TPR during prolonged therapy has been attributed to several factors including changes in the ionic content of vascular smooth muscle cells, altered ion gradients across smooth muscle cells and/or potassium channel activation, and changes in membrane-bound ATPase activity [28, 52]. The ability of thiazide-type diuretics to reduce BP seems to be linked to the presence of some level of renal function; thus, these drugs do not reduce BP in patients undergoing maintenance hemodialysis [25].
A mechanistic understanding of both diuretic action and the counterregulatory forces triggered by diuresis provide for a well-reasoned approach to the treatment of hypertension. The early action of diuretics to reduce ECF volume is optimized if dietary Na+ is restricted at the onset of therapy. This limits the repercussions of the braking phenomenon, which is an inevitable occurrence with uninterrupted diuretic use [11]. Some limitation in dietary Na+ intake may also be relevant to how diuretics might chronically reduce TPR. It is thought that intracellular Na+/Ca++ content is favorably adjusted in vascular smooth muscle cells with the acute volume reduction observed during the first several days of thiazide diuretic therapy. How the development of volume contraction specifically translates into a reduction in TPR remains unclear [28, 52]. Whatever the mechanism, it can be quite long lived, because a residual BP reduction can be seen several weeks after the withdrawal of thiazide diuretics (even without interposing non-pharmacologic treatments for maintenance of BP control) [53]. This residual BP-reducing effect upon cessation of thiazide-type diuretics has not been specifically compared to that observed with non-diuretic antihypertensive drug classes in the same patient populations.
38.10 Diuretic Class Effect
The concept of class effect has been applied to both loop diuretics and thiazide-type diuretics in respect to the management of hypertension. The loop diuretic effect on BP is a function of at least two processes: first, the manner in which volume removal is effected and, second, the capacity of these compounds to independently reduce total peripheral resistance. Small doses of the long-acting loop diuretic, torsemide, may cause significant BP reduction in essential hypertensives, a process independent of the level of diuresis, and one not seen with sub-diuretic doses of furosemide [54]. Intra-arterially infused furosemide does not directly dilate human forearm arterial vessels even at supratherapeutic concentrations [55]; in bioequivalent doses however, furosemide is just as effective as torsemide in reducing 24-h ambulatory BP in stage II–III CKD patients [56]. Until comparison studies amongst loop diuretics are conducted in diverse populations, it is premature to presume that these compounds are distinguishable, independent of volume removal, in their ability to reduce BP.
The idea of class effect for thiazide-type diuretics is still advanced by some, but it has minimal experimental support. Much of the recent debate on thiazide-type diuretic class effect has centered on the similarities and differences between chlorthalidone and HCTZ [57]. The concept of class effect with thiazide-type diuretics should be considered in two ways: first the effect on BP and second the event rate reduction. These two compounds are fundamentally different diuretics, in that chlorthalidone has a considerably longer duration of diuretic action than HCTZ [58].
One study evaluated chlorthalidone 25 mg/day compared to HCTZ 50 mg/day. In spite of the lower dose of chlorthalidone, clinic BP was reduced to a similar degree as the higher dose of HCTZ. Notably, nighttime BP was significantly lower with chlorthalidone, which is likely related to its longer duration of action [58]. A recent meta-analysis further compared the effects of chlorthalidone and HCTZ on systolic BP and serum K+ and found that chlorthalidone had a greater effect on BP with similar effects on serum K+ at equivalent doses [59]. As to the issues of outcomes, chlorthalidone has been used in several of the major clinical trials in the United States and has had a more consistent pattern of favorable outcomes than is the case with HCTZ [60–62]. These more favorable outcomes with chlorthalidone could be due to a greater reduction in nighttime BP and/or a reduction in the early morning surge in BP [29].
38.11 Clinical Usage
38.11.1 Indications
Thiazide-type diuretics are indicated in the treatment of hypertension, and they provide wide-ranging CV benefits. When used alone in the nonedematous patient, thiazide diuretics are as effective as most other antihypertensive drug classes, an observation which is independent of body mass index [63]. The degree to which diuretics lower BP relates, in part, to the level of counterregulatory system activation including increased heart rate and activation of the renin-angiotensin-aldosterone and sympathetic nervous systems. Head-to-head comparisons amongst the various thiazide-type diuretics have not shown significant differences for BP reduction when equivalent doses are used. The exception to this may be with chlorthalidone, which at a dose of 25 mg is comparatively more potent than 50 mg of HCTZ, particularly as related to overnight BP reduction [58]. Loop diuretics are less effective than thiazide-type drugs in reducing BP in the nonedematous patient [64]; however, as CKD transitions from stage 3 to 5, particularly with extracellular fluid (ECF) volume expansion, loop diuretic therapy becomes the preferred diuretic therapy for management of hypertension [65].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


